Гены контролирующие синтез гемоглобина
Содержание статьи
Гемоглобин А — Hemoglobin A
Структура гемоглобина взрослого человека. Субъединицы α и β показаны красным и синим, а железосодержащие гемовые группы — зеленым. Из PDB : 1GZX Proteopedia Hemoglobin.
Гемоглобин A (HbA), также известный как взрослый гемоглобин, гемоглобин A1 или α 2 β 2 , является наиболее распространенным тетрамером человеческого гемоглобина , составляющим более 97% от общего гемоглобина эритроцитов . Гемоглобин — это связывающий кислород белок, содержащийся в эритроцитах , который переносит кислород из легких в ткани. Гемоглобин А является наиболее распространенной взрослой формой гемоглобина и существует в виде тетрамера, содержащего две альфа-субъединицы и две бета-субъединицы (α2β2). Гемаглобин А2 (HbA2) является менее распространенной формой гемоглобина у взрослых и состоит из двух альфа и двух дельта-глобиновых субъединиц. Этот гемоглобин составляет 1-3% гемоглобина у взрослых.
Структура и функции
Гемоглобин A (HbA) является наиболее распространенной формой гемоглобина у взрослых и существует в виде тетрамера, содержащего две альфа-субъединицы и две бета-субъединицы (α2β2). Каждая субъединица содержит группу гема, с которой могут связываться молекулы двухатомного кислорода (O 2 ). Помимо кислорода, сборка субъединиц и четвертичная структура, как известно, играют важную роль в сродстве Hb. Когда гемоглобин связывается с O2 ( оксигемоглобином ), он присоединяется к железу II (Fe2 +) гема, и именно этот ион железа может связывать и расщеплять кислород для транспортировки кислорода по всему телу. Все субъединицы должны присутствовать, чтобы гемоглобин мог улавливать и выделять кислород в нормальных условиях.
Синтез
Биосинтез гема, который включает множество ферментативных стадий, которые начинаются в митохондрии и заканчиваются в цитоплазме клетки.
Синтез гема
Синтез гема включает серию ферментативных стадий, которые происходят в митохондриях и цитозоле клетки. Во-первых, в митохондрии происходит конденсация сукцинил-КоА и глицина под действием АЛК-синтазы с образованием 5-аминолевулиновой кислоты (АЛК). Затем ALA перемещается в цитозоль и после серии реакций создает копропорфиринген III . Эта молекула возвращается в митохондрию, где она реагирует с оксидазой протопорфирин-III с образованием протопорфирина IX . Затем железо ферментативно вставляется в протопорфирин через феррохелатазу с образованием гема.
Синтез глобина
Синтез глобина происходит в рибосомах , расположенных внутри цитозоля. Две цепи глобина, содержащие гемовые группы, объединяются, образуя гемоглобин. Одна из цепей является альфа-цепью, а другая — не-альфа-цепью. Природа не-альфа-цепей в молекулах гемоглобина варьируется в зависимости от различных переменных. У плода есть не-альфа-цепь, называемая гамма, а после рождения она называется бета. Бета-цепочка будет соединена с альфа-цепочкой. Это соединение двух альфа и не альфа цепей, которые создают молекулу гемоглобина. Две альфа- и две гамма-цепи образуют гемоглобин плода или гемоглобин F (HbF). По прошествии первых пяти-шести месяцев после рождения при объединении двух альфа-цепей и двух бета-цепей образуется гемоглобин взрослого человека (HbA). Гены, кодирующие альфа-цепи, расположены на хромосоме 16 , в то время как гены, кодирующие не-альфа-цепи, расположены на хромосоме 11 .
Клиническое значение
Из-за многочисленных этапов и процессов синтеза гемоглобина существует множество мест, в которых могут возникать ошибки. В синтезе гема участвует множество ферментов, и когда эти ферменты недостаточны или не функционируют должным образом, могут возникать такие последствия, как мутации или делеции в генах, кодирующих глобиновую цепь. Это приводит к нарушениям генов глобина ( гемоглобинопатиям ), которые могут быть либо аномальными вариантами цепи глобина ( серповидноклеточная анемия ), либо сниженным синтезом цепи в эритроидных клетках ( талассемия ) во время клеточного процесса кроветворения . Эти гемоглобинопатии часто наследуются по аутосомно-рецессивным признакам.
Альфа-талассемия
Альфа-талассемия (альфа-талассемия) определяется отсутствием продукции цепи альфа-глобина в гемоглобине, и те, кто несет мутацию, влияющую на цепь альфа-глобина только на одной хромосоме , считаются больными «тихой» альфа-талассемией, тогда как , если мутация присутствует на обоих, это считается признаком α-талассемии. α-талассемия чаще всего встречается в субтропических и тропических регионах, где люди, несущие этот ген, составляют 80-90% населения. Как и другие заболевания, связанные с гемоглобином (серповидно-клеточная анемия и β-талассемия), предполагается, что α-талассемия выбирается для популяций из-за того, что носители лучше защищены от малярии falciparum . Большинство носителей α-талассемии протекает бессимптомно и диагностируется, если она обнаруживается после обычных гематологических анализов или до обследования при рождении. Единичные носители гена α-глобина обычно не испытывают сильной усталости или анемии, поскольку у них есть компенсирующее увеличение количества микроцитарных эритроцитов. Напротив, у носителей легкой степени альфа-талассемии могут быть симптомы анемии из-за других факторов, не связанных конкретно с заболеванием: неправильного питания, падения уровня гемоглобина из-за кровопотери или других заболеваний.
Самая тяжелая форма альфа-талассемии — это состояние, которое начинается в младенчестве, при котором нет экспрессии альфа-генов и приводит к большому производству гемоглобина Барта (Hb Bart’s) . Наиболее частой причиной гемоглобина Барта является наследование делеционного аллеля, в котором отсутствуют функциональные гены альфа-глобина от обоих родителей. Hb Bart’s представляет собой тетрамер из четырех субъединиц гамма-глобулина и неэффективен для транспортировки кислорода к тканям из-за его очень высокого сродства к кислороду. Обычно это приводит к фатальной водянке плода и сопутствующим симптомам, включая внутриматочную анемию, замедление роста мозга, отек , деформации скелета и сердечно-сосудистые деформации, которые могут привести к сердечной недостаточности .
Бета-талассемия
Бета-талассемия (β-талассемия) — это наследственная мутация гена β-глобулина, которая вызывает снижение синтеза β-глобиновой цепи гемоглобина. Большинство мутаций представляют собой точечные мутации, которые влияют на трансляцию , контроль транскрипции и сплайсинг гена β гемоглобина и продукта гена. Считается, что лица с одной генной мутацией ( гетерозиготность ) имеют малую β-талассемию (носитель или признак β-талассемии), в то время как тем, у кого есть две генные мутации ( гомозиготность или сложная гетерозиготность), диагностируется β-талассемия или промежуточная среда. Из-за недостатка бета-глобина происходит накопление субъединиц альфа-глобина и альфа-тетрамеров, что приводит к повреждению эритроцитов. Люди азиатского, ближневосточного и средиземноморского происхождения гораздо чаще страдают β-талассемией. Было установлено, что фенотипы и генотипы заболевания сильно различаются из-за того, что в гене бета-глобина было обнаружено более 200 различных мутаций, связанных с талассемией. Людям с большой β-талассемией обычно требуется медицинская помощь в течение первых 2 лет жизни, а также регулярные переливания крови, чтобы выжить. Пациенты, у которых заболевание появляется позже, обычно не нуждаются в переливании крови, и у них диагностируется промежуточная талассемия.
A : Нормальные эритроциты показаны свободно текущими в кровеносном сосуде в верхней части диаграммы. На вставном изображении показано поперечное сечение нормального эритроцита с нормальным гемоглобином. B : Демонстрирует аномальные серповидные эритроциты, блокирующие кровоток в кровеносных сосудах (вазоокклюзионный криз ). На вставке показано поперечное сечение серповидноклеточной анемии с серповидным гемоглобином. Источник: https://www.nhlbi.nih.gov/
здоровье / темы здоровья / темы / sca /
Серповидно-клеточная анемия
Серповидный гемоглобин (HbS) является наиболее распространенным вариантом гемоглобина и возникает из-за замены аминокислоты в субъединице бета-глобина в шестом остатке от глутаминовой кислоты на валин . Существуют разные формы серповидно-клеточной анемии . HB SS, который является наиболее распространенной и тяжелой формой серповидноклеточной анемии. Hb SC возникает из-за наследования Hb S от одного родителя и Hb C ( гемоглобина C ) от другого родителя. Hb S бета-талассемия встречается реже и встречается у пациентов, унаследовавших гемоглобин бета-талассемии от одного родителя и HbS от другого. Кроме того, есть серповидно-клеточный признак (HbAS), который определяется наличием HbA и HbS. Это делает особь гетерозиготной по серповидным клеткам. По оценкам, около 300 миллионов человек в мире страдают серповидно-клеточной анемией, и около 100 миллионов из них проживают в Африке к югу от Сахары. Также наблюдается более высокая распространенность серповидно-клеточного синдрома в районах, где обычно встречается малярия, при этом распространенность в некоторых частях Африки и Саудовской Аравии достигает 25% и 60%, соответственно. Люди с HbAS имеют около 40% HbS, 56% HBA и обычно протекают бессимптомно, за исключением случаев острой нехватки кислорода в организме (гипоксии), которая может привести к симптомам серповидно-клеточной анемии. Однако HbAS не вызывает вазоокклюзионного криза, который, как известно, связан с серповидно-клеточной анемией.
Пациенты, гомозиготные по HbS, имеют многонитевые волокна, которые вызывают изменение формы эритроцитов от двояковогнутых дисков до удлиненных полумесяцев. Серповидная реакция обратима после повторного насыщения кислородом гемоглобина, поэтому красные кровяные тельца могут проходить циклы серповидного и некорпусного действия в зависимости от концентрации кислорода, присутствующего в кровотоке. Серповидные эритроциты не обладают гибкостью и прилипают к стенкам кровеносных сосудов, уменьшая или останавливая приток кислорода к близлежащим тканям. Это уменьшение кислорода в тканях вызывает вазоокклюзионный кризис, который проявляется в мышечной боли и повреждении тканей. Некоторые симптомы серповидно-клеточной анемии включают лихорадку, усталость от анемии , отек рук и ног, инсульт и органную недостаточность. Текущие методы лечения включают переливание крови, которое помогает увеличить количество нормальных эритроцитов, трансплантацию костного мозга, чтобы помочь организму пациента производить здоровые эритроциты, и лекарства, которые помогают облегчить симптомы, перечисленные ранее.
Смотрите также
Источник
Биосинтез гемоглобина
Учитывая, что белковая часть молекулы гемоглобина (глобин) синтезируется, как и все остальные белки, далее подробно рассмотрен биосинтез его простетической группы, т.е. синтез тетрапиррольного соединения — гема (см. главу 2).
К настоящему времени почти полностью выяснены основные пути образования порфиринов и протопорфиринов, являющихся непосредственными предшественниками гема и хлорофилла. Благодаря исследованиям Д. Шемина и др. выяснены основные пути синтеза гема. С помощью меченых предшественников было показано, что в синтезе гема в бесклеточных экстрактах эритроцитов птиц специфическое участие принимают глицин, уксусная и янтарная кислоты. Источником всех 4 атомов азота и 8 атомов углерода тетрапиррольного кольца оказался глицин, а источником остальных 26 из 34 атомов углерода — янтарная кислота (сук-цинат), точнее ее производное сукцинил-КоА. Последовательность химических реакций синтеза тетрапирролов в организме животных можно условно разделить на несколько стадий.
На I стадии, протекающей в 2 этапа, сукцинил-КоА взаимодействует с глицином и образованием δ-аминолевулиновой кислоты (δ-АЛК).
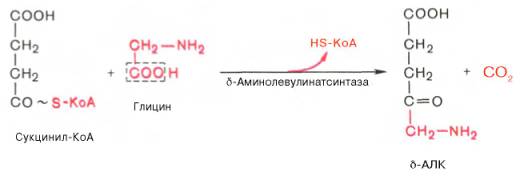
Эту стадию катализирует специфический пиридоксальфосфатзависимый фермент δ-аминолевулинатсинтаза — ключевой, аллостерический фермент синтеза тетрапирролов.
Впервые эта синтаза была обнаружена в эндоплазматической сети клеток печени. Фермент индуцируется стероидами и другими факторами и ингибируется по типу обратной связи конечным продуктом биосинтеза — гемом.
На II стадии происходит конденсация 2 молекул δ-аминолевулиновой кислоты с образованием первого монопиррольного соединения — порфо-билиногена (ПБГ).
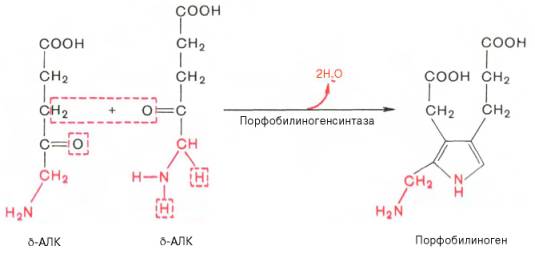
Фермент, катализирующий эту стадию,- порфобилиногенсинтаза также является регуляторным ферментом, подвергаясь ингибированию конечными продуктами синтеза. Предполагают, что механизм этой сложной реакции дегидратации включает образование кетиминной связи (шиффово основание) между кетогруппой одной молекулы δ-аминолевулиновой кислоты и δ-аминогруппой лизина молекулы фермента. В следующей многоступенчатой стадии, катализируемой соответствующими ферментами, из 4 монопиррольных молекул порфобилиногена синтезируется тетра-пиррольный комплекс протопорфирин IX, являющийся непосредственным предшественником гема. Некоторые этапы сложного пути синтеза окончательно не установлены.
В заключительной стадии протопорфирин IX присоединяет молекулу железа при участии феррохелатазы (гемсинтазы), и образуется гем. Последний используется для биосинтеза всех гемсодержащих хромопро-теинов.
Источником железа для этой реакции является ферритин, который считается резервным гемопротеином, откладывающимся в клетках костного мозга, печени и селезенки.
Имеются указания, что, помимо железа, в синтезе гема участвуют некоторые кофакторы, в частности витамин В12, ионы меди, хотя конкретная их роль не раскрыта.
Таким образом, весь путь синтеза гема может быть представлен в виде схемы, в которой даны полные и сокращенные обозначения промежуточных метаболитов и ферментов.
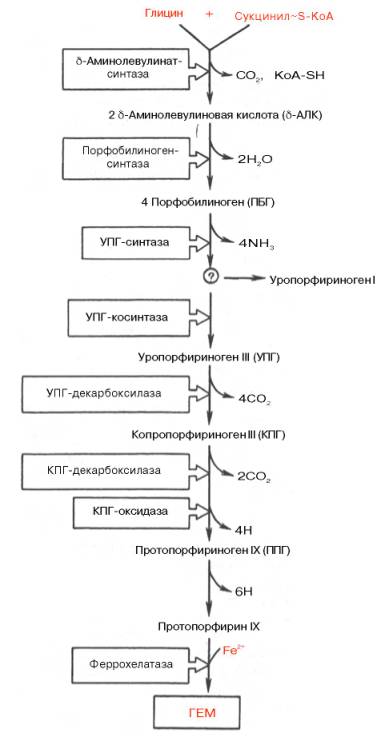
Источник
Гемоглобин
Молекула гемоглобина: 4 субъединицы окрашены в разные цвета
Структура гемоглобина человека. Железосодержащие гем-группы показаны зелёным. Красным и синим показаны альфа- и бета- субъединицы.
Гемоглоби́н (от др.-греч. αἷμα «кровь» + лат. globus «шар») (Hb или Hgb) — сложный железосодержащий белок животных, обладающих кровообращением, способный обратимо связываться с кислородом, обеспечивая его перенос в ткани. У позвоночных животных содержится в эритроцитах, у большинства беспозвоночных растворён в плазме крови (эритрокруорин) и может присутствовать в других тканях[1]. Молекулярная масса гемоглобина человека — около 66,8 кДа. Молекула гемоглобина может нести до четырёх молекул кислорода. Один грамм гемоглобина может переносить до 1,34 мл кислорода.
Гемоглобин появился более чем 400 миллионов лет назад у последнего общего предка человека и акул в результате 2 мутаций, приведших к формированию четырёхкомпонентного комплекса гемоглобина, сродство которого к кислороду достаточно для связывания кислорода в насыщенной им среде, но недостаточно, чтобы удерживать его в других тканях организма.[2][3]
Большой вклад в исследование структуры и функционирования гемоглобина внёс Макс Фердинанд Перуц, получивший за это в 1962 году Нобелевскую премию[4].
Нормальным содержанием гемоглобина в крови человека считается: у мужчин — 130-160 г/л (нижний предел — 120, верхний предел — 180 г/л), у женщин — 120-160 г/л; у детей нормальный уровень гемоглобина зависит от возраста и подвержен значительным колебаниям. Так, у детей через 1-3 дня после рождения нормальный уровень гемоглобина максимален и составляет 145-225 г/л, а к 3-6 месяцам снижается до минимального уровня — 95-135 г/л, затем с 1 года до 18 лет отмечается постепенное увеличение нормального уровня гемоглобина в крови[5].
Во время беременности в организме женщины происходит задержка и накопление жидкости, что является причиной гемодилюции — физиологического разведения крови. В результате наблюдается относительное снижение концентрации гемоглобина (при беременности уровень гемоглобина в норме составляет 110-155 г/л). Кроме этого, в связи с внутриутробным ростом ребёнка происходит быстрое расходование запасов железа и фолиевой кислоты. Если до беременности у женщины был дефицит этих веществ, проблемы, связанные со снижением гемоглобина, могут возникнуть уже на ранних сроках беременности[6].
Главные функции гемоглобина: перенос кислорода и буферная функция. У человека в капиллярах лёгких в условиях избытка кислорода последний соединяется с гемоглобином. Потоком крови эритроциты, содержащие молекулы гемоглобина со связанным кислородом, доставляются к органам и тканям, где кислорода мало; здесь необходимый для протекания окислительных процессов кислород освобождается от связи с гемоглобином. Кроме того, гемоглобин способен связывать в тканях небольшое количество диоксида углерода (CO2) и освобождать его в лёгких.
Монооксид углерода (CO) связывается с гемоглобином крови намного сильнее (в 250 раз[7]), чем кислород, образуя карбоксигемоглобин (HbCO). Впрочем, монооксид углерода может быть частично вытеснен из гема при повышении парциального давления кислорода в лёгких. Некоторые процессы приводят к окислению иона железа в гемоглобине до степени окисления +3. В результате образуется форма гемоглобина, известная как метгемоглобин (HbOH) (metHb, от «мета-» и «гемоглобин», иначе гемиглобин или ферригемоглобин, см. Метгемоглобинемия). В обоих случаях блокируются процессы транспортировки кислорода.
Строение[править | править код]
Гемоглобин является сложным белком класса гемопротеинов, то есть в качестве простетической группы здесь выступает гем — порфириновое ядро, содержащее железо. Гемоглобин человека является тетрамером, то есть состоит из 4 протомеров. У взрослого человека они представлены полипептидными цепями α1, α2, β1 и β2. Субъединицы соединены друг с другом по принципу изологического тетраэдра. Основной вклад во взаимодействие субъединиц вносят гидрофобные взаимодействия. И α-, и β-цепи относятся к α-спиральному структурному классу, так как содержат исключительно α-спирали. Каждая цепь содержит восемь спиральных участков, обозначаемых буквами от A до H (от N-конца к C-концу).
Гем представляет собой комплекс протопорфирина IX, относящегося к классу порфириновых соединений, с атомом железа(II). Этот кофактор нековалентно связан с гидрофобной впадиной молекул гемоглобина и миоглобина.
Железо(II) характеризуется октаэдрической координацией, то есть связывается с шестью лигандами. Четыре из них представлены атомами азота порфиринового кольца, лежащими в одной плоскости. Две другие координационные позиции лежат на оси, перпендикулярной плоскости порфирина. Одна из них занята азотом остатка гистидина в 93-м положении полипептидной цепи (участок F). Связываемая гемоглобином молекула кислорода координируется к железу с обратной стороны и оказывается заключённой между атомом железа и азотом ещё одного остатка гистидина, располагающегося в 64-м положении цепи (участок E).
Всего в гемоглобине человека четыре участка связывания кислорода (по одному гему на каждую субъединицу), то есть одновременно может связываться четыре молекулы. Гемоглобин в лёгких при высоком парциальном давлении кислорода соединяется с ним, образуя оксигемоглобин. При этом кислород соединяется с гемом, присоединяясь к железу гема на 6-ю координационную связь. На эту же связь присоединяется и монооксид углерода, вступая с кислородом в «конкурентную борьбу» за связь с гемоглобином, образуя карбоксигемоглобин.
Связь гемоглобина с монооксидом углерода более прочная, чем с кислородом. Поэтому часть гемоглобина, образующая комплекс с монооксидом углерода, не участвует в транспорте кислорода. В норме у человека образуется 1,2 % карбоксигемоглобина. Повышение его уровня характерно для гемолитических процессов, в связи с этим уровень карбоксигемоглобина является показателем гемолиза.
Физиология[править | править код]
Изменение состояний окси- и дезоксигемоглобина
В отличие от миоглобина гемоглобин имеет четвертичную структуру, которая придаёт ему способность регулировать присоединение и отщепление кислорода и характерную кооперативность: после присоединения первой молекулы кислорода связывание последующих облегчается. Структура может находиться в двух устойчивых состояниях (конформациях): оксигемоглобин (содержит 4 молекулы кислорода; напряжённая конформация) и дезоксигемоглобин (кислорода не содержит; расслабленная конформация).
Устойчивое состояние структуры дезоксигемоглобина усложняет присоединение к нему кислорода. Поэтому для начала реакции необходимо достаточное парциальное давление кислорода, что возможно в альвеолах лёгких. Изменения в одной из 4 субъединиц влияют на оставшиеся, и после присоединения первой молекулы кислорода связывание последующих облегчается.
Отдав кислород тканям, гемоглобин присоединяет к себе ионы водорода и углекислый газ, перенося их в лёгкие[8].
Гемоглобин является одним из основных белков, которыми питаются малярийные плазмодии — возбудители малярии, и в эндемичных по малярии районах земного шара весьма распространены наследственные аномалии строения гемоглобина, затрудняющие малярийным плазмодиям питание этим белком и проникновение в эритроцит. В частности, к таким имеющим эволюционно-приспособительное значение мутациям относится аномалия гемоглобина, приводящая к серповидноклеточной анемии. Однако, к несчастью, эти аномалии (как и аномалии строения гемоглобина, не имеющие явно приспособительного значения) сопровождаются нарушением кислород-транспортирующей функции гемоглобина, снижением устойчивости эритроцитов к разрушению, анемией и другими негативными последствиями. Аномалии строения гемоглобина называются гемоглобинопатиями.
Гемоглобин высокотоксичен при попадании значительного его количества из эритроцитов в плазму крови (что происходит при массивном внутрисосудистом гемолизе, геморрагическом шоке, гемолитических анемиях, переливании несовместимой крови и других патологических состояниях). Токсичность гемоглобина, находящегося вне эритроцитов, в свободном состоянии в плазме крови, проявляется тканевой гипоксией — ухудшением кислородного снабжения тканей, перегрузкой организма продуктами разрушения гемоглобина — железом, билирубином, порфиринами с развитием желтухи или острой порфирии, закупоркой почечных канальцев крупными молекулами гемоглобина с развитием некроза почечных канальцев и острой почечной недостаточности.
Ввиду высокой токсичности свободного гемоглобина в организме существуют специальные системы для его связывания и обезвреживания. В частности, одним из компонентов системы обезвреживания гемоглобина является особый плазменный белок гаптоглобин, специфически связывающий свободный глобин и глобин в составе гемоглобина. Комплекс гаптоглобина и глобина (или гемоглобина) затем захватывается селезёнкой и макрофагами тканевой ретикуло-эндотелиальной системы и обезвреживается.
Другой частью гемоглобинообезвреживающей системы является белок гемопексин[en], специфически связывающий свободный гем и гем в составе гемоглобина. Комплекс гема (или гемоглобина) и гемопексина затем захватывается печенью, гем отщепляется и используется для синтеза билирубина и других жёлчных пигментов или выпускается в рециркуляцию в комплексе с трансферринами для повторного использования костным мозгом в процессе эритропоэза.
Экспрессия генов гемоглобина до и после рождения.
Также указаны типы клеток и органы, в которых происходит экспрессия гена (данные по Wood W. G., (1976). Br. Med. Bull. 32, 282.).[9]
Гемоглобин при заболеваниях крови[править | править код]
Дефицит гемоглобина может быть вызван, во-первых, уменьшением количества молекул самого гемоглобина (см. анемия), во-вторых, из-за уменьшенной способности каждой молекулы связать кислород при том же самом парциальном давлении кислорода.
Гипоксемия — это уменьшение парциального давления кислорода в крови, её следует отличать от дефицита гемоглобина. Хотя и гипоксемия, и дефицит гемоглобина являются причинами гипоксии. Если дефицит кислорода в организме в общем называют гипоксией, то местные нарушения кислородоснабжения называют ишемией.
Прочие причины низкого гемоглобина разнообразны: кровопотеря, пищевой дефицит, болезни костного мозга, химиотерапия, отказ почек, атипичный гемоглобин.
Повышенное содержание гемоглобина в крови связано с увеличением количества или размеров эритроцитов, что наблюдается также при истинной полицитемии. Это повышение может быть вызвано: врождённой болезнью сердца, лёгочным фиброзом, слишком большим количеством эритропоэтина.
См. также[править | править код]
- Гемоглобин А
- Гемоглобин С (мутантная форма)
- Эмбриональный гемоглобин
- Гемоглобин S (мутантная форма)
- Гемоглобин F (фетальный)
- Кобоглобин
- Нейроглобин
- Анемия
- Порфирия
- Талассемия
- Эффект Вериго — Бора
Примечания[править | править код]
- ↑ Haemoglobins of invertebrate tissues. Nerve haemoglobins of Aphrodite, Aplysia and Halosydna
- ↑ Ученые выяснили происхождение гемоглобина. РИА Новостей, 20.05.2020, 18:59
- ↑ Michael Berenbrink. Evolution of a molecular machine/Nature, NEWS AND VIEWS, 20 MAY 2020
- ↑ Лауреаты нобелевской премии. Макс Перуц.
- ↑ Назаренко Г. И., Кишкун А. А. Клиническая оценка результатов лабораторных исследований. — 2005.
- ↑ Общий анализ крови и беременность Архивная копия от 10 марта 2014 на Wayback Machine
- ↑ Hall, John E. Guyton and Hall textbook of medical physiology (англ.). — 12th ed.. — Philadelphia, Pa.: Saunders/Elsevier, 2010. — P. 1120. — ISBN 978-1416045748.
- ↑ Степанов В. М. Структура и функции белков : Учебник. — М. : Высшая школа, 1996. — С. 167-175. — 335 с. — 5000 экз. — ISBN 5-06-002573-X.
- ↑ Айала Ф., . Современная генетика: В 3-х т = Modern Genetics / Пер. А. Г. Имашевой, А. Л. Остермана, . Под ред. Е. В. Ананьева. — М.: Мир, 1987. — Т. 2. — 368 с. — 15 000 экз. — ISBN 5-03-000495-5.
Литература[править | править код]
- Mathews, CK; van Holde, KE & Ahern, KG (2000), Biochemistry (3rd ed.), Addison Wesley Longman, ISBN 0-8053-3066-6
- Levitt, M & Chothia, C (1976), Structural patterns in globular proteins, Nature
Ссылки[править | править код]
- Eshaghian, S; Horwich, TB; Fonarow, GC (2006). «An unexpected inverse relationship between HbA1c levels and mortality in patients with diabetes and advanced systolic heart failure». Am Heart J. 151 (1): 91.e1-91.e6. DOI:10.1016/j.ahj.2005.10.008. PMID 16368297.
- Kneipp J, Balakrishnan G, Chen R, Shen TJ, Sahu SC, Ho NT, Giovannelli JL, Simplaceanu V, Ho C, Spiro T (2005). «Dynamics of allostery in hemoglobin: roles of the penultimate tyrosine H bonds». J Mol Biol. 356 (2): 335-53. DOI:10.1016/j.jmb.2005.11.006. PMID 16368110.
- Hardison, Ross C. (2012). «Evolution of Hemoglobin and Its Genes». Cold Spring Harbor Perspectives in Medicine. 2 (12): a011627. DOI:10.1101/cshperspect.a011627. ISSN 2157-1422. PMC 3543078. PMID 23209182.
Источник