Механизм проникновения холестерина в эндотелий сосудов
Содержание статьи
Механизмы развития атеросклероза — bariatric.1spbgmu.ru
МЕХАНИЗМЫ РАЗВИТИЯ АТЕРОСКЛЕРОЗА ПРИ ОЖИРЕНИИ
В настоящее время атеросклероз рассматривается как системное заболевание, характеризующееся формированием одиночных и множественных атероматозных бляшек (очагов липидных, главным образом – холестериновых, отложений) на внутренней поверхности крупных артерий, приводящих к нарушению их проходимости. Изменения структуры стенок сосудов в результате отложения холестерина стимулирует процессы локального воспаления, сопровождающиеся постепенным развитием соединительной ткани (склерозом) и отложением солей кальция (кальцинозом), существенно усугубляющими сужение просвета артерий и нарушение тока крови по ним.
Термин “атеросклероз” (“athtre” – пшеничная каша, “sclerosis” — твердый) предложен в 1904 году F. Marchand. Этиология (причины) и патогенез (процесс развития) заболевания исключительно сложны и не до конца изучены. Отмеченное обстоятельство объясняет повышенный интерес к проблеме не только среди специалистов практической и теоретической медицины, но и исследователей, работающих в различных областях фундаментальной науки: химии, физики, биологии и др. Изучение тонких механизмов развития атеросклероза неразрывно связано с пониманием таких глубоких и принципиальных вопросов функционирования живых многоклеточных организмов, как передача наследственной информации, старение, развитие системных заболеваний и злокачественных новообразований и многих других нерешенных задач человечества.
Значительное количество “белых пятен” в современных представлениях о проблеме является причиной существования конкурирующих взглядов на механизмы возникновения и прогрессирования атеросклероза, имеющих свои сильные и слабые стороны, однако, в общих чертах, укладывающихся в две основные концепции.
Нарушения процессов регуляции метаболизма липидов (дислипидемий), и в первую очередь — обмена холестерина, составляют основу “инфильтративно-гиперпластической” или “холестериновой” теории (Н. Н. Аничкова и С.С. Халатова). В течении века существования данной концепции первоначальные теоретические положения получили убедительные подтверждения ее научной обоснованности и стали базой для дальнейшего изучения механизмов развития заболевания с позиций современных знаний.
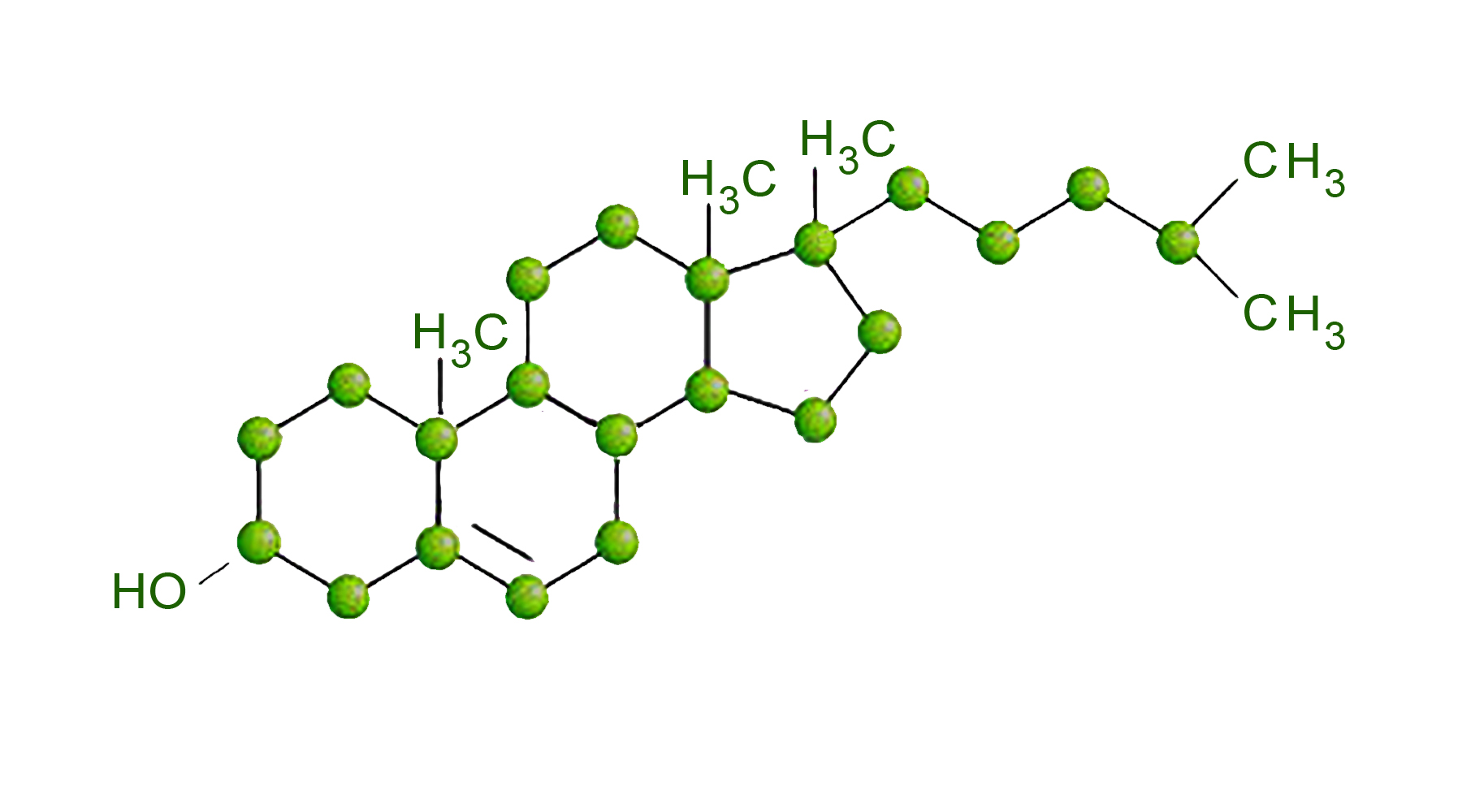
Строение молекулы холестерина
К настоящему времени получено множество доказательств, что липиды, в том числе холестерин и его эфиры, располагающиеся внутри и вокруг клеток интимы (внутренней оболочки) и медии (средней части стенки) артерий и участвующие в образовании атеросклеротических бляшек, происходят из липопротеинов плазмы крови.
Липидный спектр плазмы человека составляют триглицериды (эфиры жирных кислот и глицерина), фосфолипиды (сложные производные жирных кислот и фосфорной кислоты), эфиры холестерина (соединения жирных кислот и холестерина), а также свободные (неэстерифицированные) жирные кислоты.
Эфиры холестерина в качестве обязательного липидного компонента входят в состав липопротеинов (комплексных соединений с белками — апопротеинами), представляющих универсальный механизм транспорта и передачи тканям различных производных жирных кислот. В зависимости от состава и размеров различают липопротеины очень низкой плотности (ЛПОНП), низкой плотности (ЛПНП), высокой плотности (ЛПВП), липопротеиды средней плотности (ЛПСП) и хиломикроны. Каждая группа липопротеинов очень неоднородна по размерам частиц, а также содержанию апопротеинов и липидов. Основная часть холестерина переносится липопротеинами низкой плотности, существенно меньшая – ЛПОНП и липопротеинами высокой плотности. В отличие от холестерина, эндогенные триглицериды транспортируются преимущественно в составе ЛПОНП.
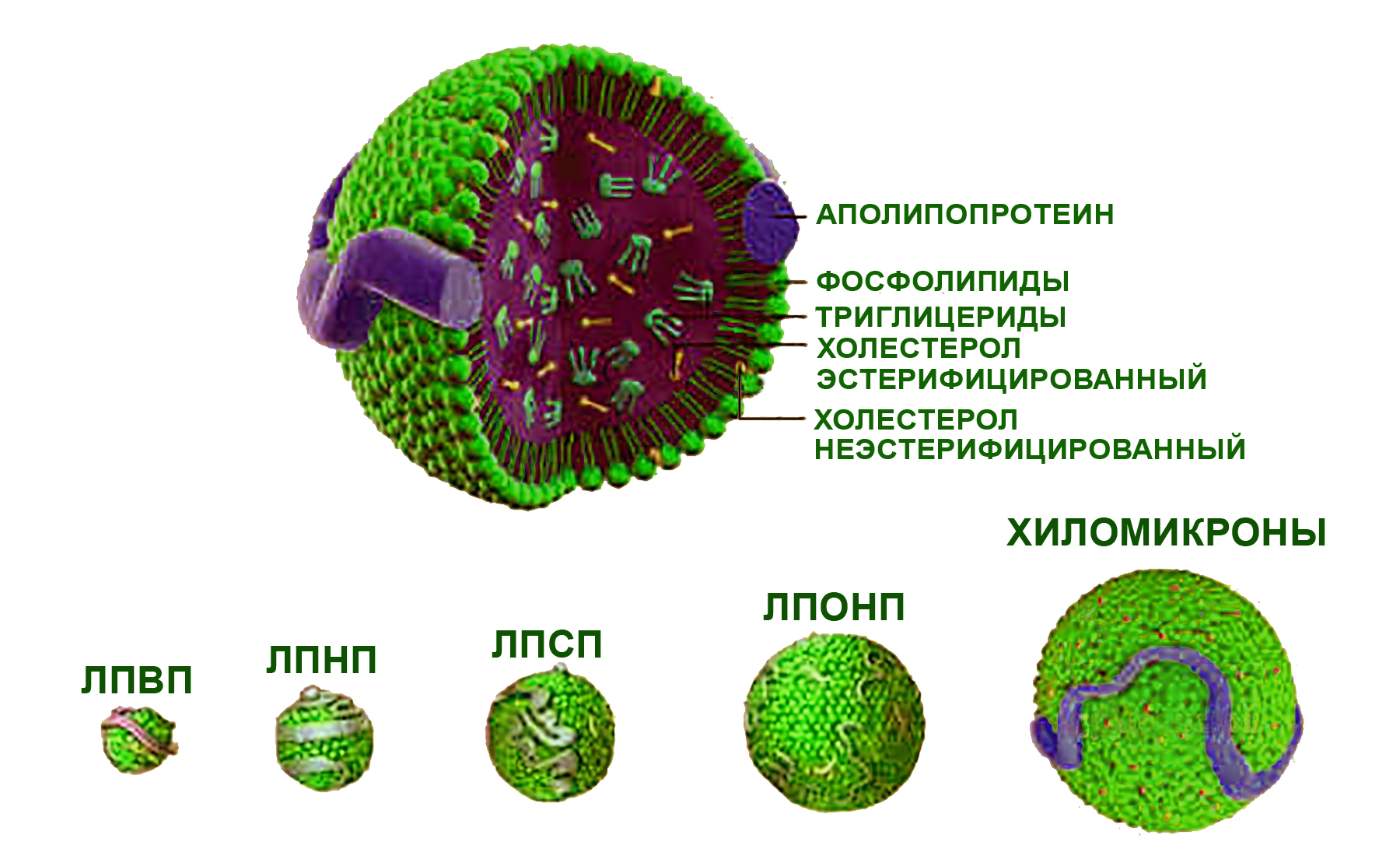
Строение и соотношение размеров липопротеинов
Функцией апопротеинов, входящих в состав липопротеинов, являются повышение растворимости (в плазме крови) эфиров холестерина и триглицеридов, регуляция и кооперация взаимодействия липидов с ферментами (в процессе биохимических реакций) и связь с рецепторами при транспорте соединений жирных кислот в клетки тканей.
С позиций современных знаний внутриклеточный и тканевой метаболизм липопротеинов разных классов упрощенно можно представить следующим образом. Хиломикроны, образующиеся в тонкой кишке из липидов пищи, поступают через лимфу в плазму крови, собирающуюся через систему воротной вены печенью. Под действием ферментов плазмы крови (липопротеинлипазы) хиломикроны распадаются на более мелкие комплексы (ремнанты), которые и захватываются гепатоцитами (клетками печени). Синтезированные в печени триглицериды в составе ЛПОНП поступают в плазму, где они, как и хиломикроны, претерпевают частичную деградацию до ремнантных липопротеинов низкой и особо низкой плотности. Циркулирующие в плазме ЛППП (липопротеины промежуточной плотности) и ЛПОНП утилизируются клетками различных тканей, а при их избытке – подвергаются катаболизму (разложению) в печени до триглицеридов. Липопротеины высокой плотности образуются из холестерина, синтезированного клетками печени и других тканей, или поступившего с пищей, и компонентов (фосфолипидов) хиломикронов и ЛПНП.
Столь подробное изложение сложных процессов синтеза, транспорта, резервирования и утилизации производных жирных кислот в организме позволяет представить многогранность возможных причин нарушения механизмов их регуляции, играющих роль в развитии дислипопротеинемии (изменении концентрации различных липопротеинов в плазме крови).
Роль генетических факторов в возникновении атеросклероза в настоящее время не вызывает сомнений и доказана существованием как минимум трех описанных патологических состояний (дислипопротеинемий): семейной гиперхолестеринемии, семейной комбинированной гиперлипидемии и семейной гипертриглицеридемии.
Семейная гиперхолестеринемия является следствием мутации гена, контролирующего синтез рецепторов к ЛПНП, приводящих в итоге к повышенному синтезу холестерина и ЛПОНП. Данный вариант наследственной дислипидемии приблизительно в трети случаев характеризуется злокачественным течением атеросклероза и практически не поддается консервативной терапии.
Семейная комбинированная гиперлипидемия вызывается доминантным (подавляющим проявление другого, отвечающего за тот же признак) геном. У людей с быстрым развитием атеросклероза на долю данной мутации приходится 15% случаев.
Третьим из наиболее распространенных наследственных заболеваний, приводящих к быстрому развитию атеросклероза, является семейная гипертриглицеридемия. Мутация доминантного гена проявляется значительным повышением концентрации в плазме крови ЛПОНП и снижением уровня ЛПВП. Данный тип генетических изменений отмечается у 5% больных.
Наследственные варианты развития атеросклероза отмечаются менее чем у 10% пациентов с данным заболеванием. Большая же часть гиперлипопротеинемий относится к категории приобретенных. Причиной их возникновения является совокупность внешних факторов (характера питания, приема некоторых медикаментозных препаратов, сопутствующих заболеваний и т.д.) и его генетических особенностей (к сожалению, многочисленных и не до конца понятных). Сложный комплекс взаимодействий между указанными экзогенными (внешними) и эндогенными (внутренними) факторами может приводить к нарушению обмена липидов, и в итоге – к развитию атеросклероза.
Развитие атеросклероза зависит не только от абсолютного уровня тех или иных классов липопротеинов, но и от соотношения липопротеинов с атерогенной (вызывающей атеросклероз) и антиантиатерогенной (препятствующей возникновению заболевания) направленностью. Ярко выраженным атерогенным эффектом обладают ЛПНП и ЛПОНП, в то время как липопротеины высокой плотности препятствуют развитию заболевания.
Таковы общие представления о роли нарушений липидного обмена в возникновении и прогрессировании атеросклероза с позиций “холестериновой” теории.
Однако неверным было бы рассмотрение патогенеза данного состояния исключительно с точки зрения дисбаланса между различными липопротеинами в плазме крови, поскольку характерные для атеросклероза изменения происходят именно в стенках крупных артерий.
Начальными признаками развития заболевания являются набухание и разрыхление внутреннего слоя (эндотелия) кровеносных сосудов. Изменение свойств эндотелия (которое может вызываться повышением артериального давления, воздействием вирусов, бактерий, эндо- или эзогенных токсинов и т.д.) приводит к агрегации и адгезии (концентрации и прилипанию) тромбоцитов, являющимися начальной фазой физиологической реакции коагуляции (свертывания) крови и формирования микротромбов при нарушении целостности сосудов. Параллельно с процессом агрегации тромбоцитов происходит синтез фибрина, нити которого являются “каркасом” для клеточных элементов тромба. Продуцируемые тромбоцитами биологически активные вещества (цитокины) инициируют процесс воспаления, усиливают образование первоначального тромба и вызывают адгезию других клеточных (лимфоцитов, моноцитов, макрофагов и др.) и молекулярных компонентов плазмы крови, включая атерогенные липопротеины. Способностью к эндоцитарному (внутриклеточному) захвату частиц ЛПНП и ЛПОНП под действием медиаторов воспаления могут обладать клетки эндотелия и других слоев стенки артерий, а также — присутствующие в зоне тромба макрофаги (специфические клетки, уничтожающие в организме чужеродные агенты).
С течением времени тромбоциты, выполнившие свою физиологическую функцию, подвергаются распаду и утилизации (макрофагами), а в зоне первоначального повреждения сосуда формируется атеросклеротическая бляшка, состоящая из нитей фибрина (соединительной ткани) и отложений холестерина. Воспалительные изменения приобретают характер хронических (постоянных) вследствие повреждающего действия турбулентного (вихревого) потока крови в области нарушения структуры внутренней поверхности артериальной стенки и изменения ее естественного строения. Воспалительный процесс стимулирует гибель и заместительную пролиферацию миоцитов (клеток мышечных клеток мышечного слоя), развитие соединительной ткани, отложение солей кальция (кальциноз), в итоге приводящих к еще более выраженному локальному утолщению и разрыхлению стенки с сужением просвета сосуда.
Описанные изменения проходят ряд последовательных стадий.
- Образование липидного пятна (или полоски), представляющего собой участки бледно-желтого цвета, содержащие липиды, не возвышающиеся над поверхностью интимы артерии.
- Формирование фиброзной бляшки – овального или округлого образования, содержащего липиды, возвышающегося над поверхностью интимы и нередко сливающегося в сплошные бугристые поля.
- Кальциноз – отложение в фиброзной бляшке солей кальция.
- Деструктивные изменения фиброзной бляшки: изъязвление, кровоизлияние, наложение тромботических масс.
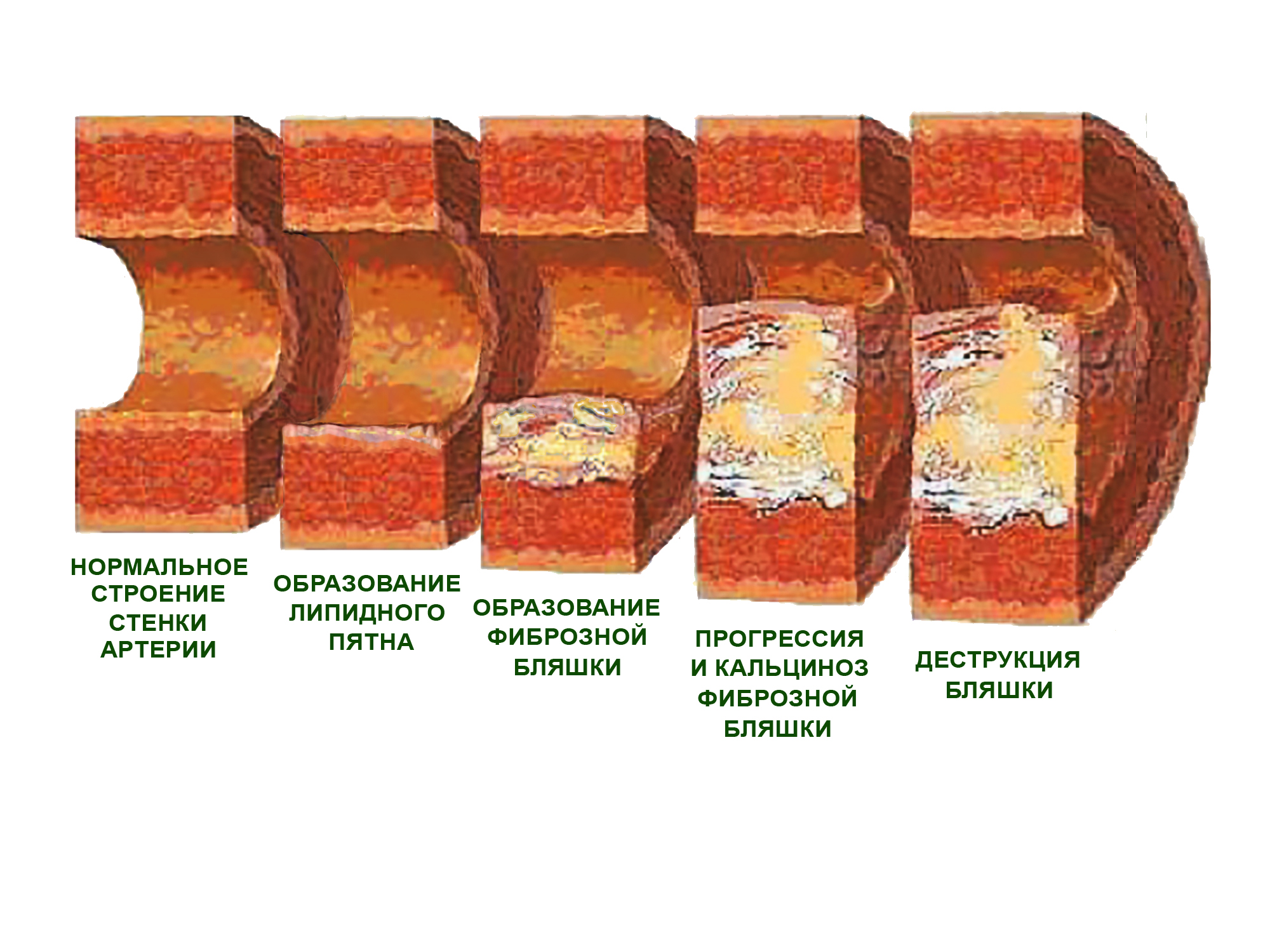
Стадии развития атеросклеротической бляшки
Таковы основные положения “эндотелиальной” концепции развития атеросклероза. Однако так же, как “холестериновая” теория, данная гипотеза имеет ряд слабых мест и не позволяет ответить на ключевой вопрос: почему, при прочих равных условиях, в одних случаях заболевание возникает, а в других – нет?
Современные представления о проблеме позволяют считать, что развитие атеросклероза происходит с участием изложенных в свете обоих теорий процессов. Вероятно, в развитии заболевания принимают участие и многие другие, пока не изученные, механизмы.
Однако даже существующие на сегодняшний день взгляды дают возможность очертить круг внешних и внутренних факторов, несущих риск возникновения атеросклероза, и сформировать комплексную систему профилактики и лечения данного системного патологического процесса и его осложнений.
Источник
ГЛАВА 01. АТЕРОСКЛЕРОЗ
ГЛАВА 01. АТЕРОСКЛЕРОЗ
Атеросклероз — патологический процесс, приводящий к изменению стенки артерий в результате накопления липидов, образования фиброзной ткани и формирования бляшки, сужающей просвет сосуда. Атеросклероз не считают самостоятельным заболеванием, клинически он проявляется общими и/или местными расстройствами кровообращения, часть из которых выделена в отдельные нозологические формы.
Наиболее часто атеросклеротический процесс развивается в аорте, бедренных, подколенных, большеберцовых, венечных, внутренней и наружной сонных артериях и артериях мозга. Атеросклеротические изменения, как правило, возникают в местах бифуркации аорты и артерий. Осложнения атеросклероза обусловливают 1/2 всех смертельных случаев и 1/3 смертельных случаев у лиц в возрасте 35-65 лет.
ЛИПИДЫ КРОВИ
В плазме крови холестерин и триглицериды связаны с белками и называются липопротеинами (ЛП). Степень их участия в атерогенезе зависит от размеров ЛП.
• Наименьшие по размеру (5-12 нм) — ЛП высокой плотности (ЛПВП) — легко проникают в стенку артерий и так же легко её покидают, не участвуя, таким образом, в атерогенезе.
• ЛП низкой плотности (ЛПНП, 18-25 нм), ЛП промежуточной плотности (ЛППП, 25-35 нм) и небольшая часть ЛП очень низкой плотности (ЛПОНП, размер около 50 нм) достаточно малы для того, чтобы проникнуть в стенку артерий. После окисления эти ЛП легко задерживаются в стенке артерий.
• Крупные по размеру ЛП — хиломикроны (75-1200 нм) и ЛПОНП значительных размеров (80 нм) — слишком велики для того, чтобы проникнуть в артерии, и поэтому не считаются атерогенными.
Между содержанием холестерина ЛПНП и риском развития ишемической болезни сердца (ИБС) имеется прямая зависимость — чем больше содержание холестерина ЛПНП, тем выше риск развития ИБС. Между содержанием холестерина ЛПВП и риском развития ИБС имеется обратная зависимость — чем выше содержание в крови холестерина ЛПВП, тем ниже риск развития ИБС.
Триглицериды содержатся в основном в хиломикронах (80-95%), они синтезируются в слизистой оболочке тонкой кишки из жиров, поступающих с пищей, и в ЛПОНП (55-80%). Выраженную гипертриглицеридемию не считают атерогенной, так как крупные по размеру хиломикроны и ЛПОНП не могут проникнуть через стенку артерий, однако значительную гипертриглицеридемию может вызвать панкреатит. ЛПНП и ЛПВП содержат небольшое количество триглицеридов (5-15%).
АТЕРОГЕНЕЗ
Патогенез атеросклеротического поражения кровеносных сосудов (атерогенез) призваны объяснить три гипотезы. Само поражение стенки сосуда развивается постадийно.
СТАДИИ АТЕРОГЕНЕЗА
Атеросклеротические изменения происходят во внутренней оболочке артерий. Этот процесс протекает в три стадии (рис. 1-1): жировая полоска, фиброзная бляшка и комплексные нарушения.
Рис. 1-1. Атеросклеротические изменения в артерии: а — жировая полоска; б — фиброзная бляшка; в — комплексные нарушения. 1 — межклеточные липиды; 2 — пенистые клетки; 3 — фиброзная капсула; 4 — гладкомышечные клетки; 5 — липидное ядро; 6 — тромб; 7 — изъязвление; 8 — кальцификация; 9 — кровоизлияние
Жировая полоска — раннее морфологическое проявление атеросклероза. С момента рождения человека в сосудах можно обнаружить пятна желтоватой окраски размером 1-2 мм. Эти пятна, представляющие собой отложения липидов, с течением времени увеличиваются и сливаются друг с другом. Гладкомышечные клетки и макрофаги появляются во внутренней оболочке артерий, макрофаги накапливают липиды и превращаются в пенистые клетки. Так возникает жировая полоска, состоящая из гладкомышечных клеток и содержащих липиды макрофагов. Но отложение липидов в виде жировых полосок в стенке артерий не означает обязательное перерастание процесса в следующую стадию (формирование фиброзной бляшки).
Фиброзная бляшка располагается во внутренней оболочке артерий и растёт эксцентрично, со временем уменьшая просвет сосуда. Фиброзная бляшка имеет плотную капсулу, состоящую из клеток эндотелия, гладкомышечных клеток, Т-лимфоцитов, пенистых клеток (макрофагов), фиброзной ткани, и мягкое ядро, содержащее эфиры и кристаллы холестерина. Холестерин образуется не за счёт локального синтеза, а поступает из крови.
Комплексные нарушения заключаются в уменьшении толщины капсулы фиброзной бляшки менее 65 мкм и нарушении её целостности — возникновении трещин, язв, разрывов. Этому способствуют следующие факторы.
• Увеличение зоны атероматоза более чем на 30-40% от общего объёма фиброзной бляшки (за счёт накопления холестерина).
• Инфильтрация поверхности фиброзной бляшки макрофагами (более 15% поверхности бляшки), приводящая к асептическому воспалению.
• Воздействие металлопротеаз, продуцируемых макрофагами и вызывающих деструкцию коллагена, эластина и гликопротеинов.
• Высокое содержание окисленных ЛПНП, вызывающих продукцию медиаторов воспаления и стимуляцию адгезии моноцитов.
Нарушение целостности фиброзной бляшки приводит к прикреплению к ней тромбоцитов, их агрегации, тромбозу и развитию клинической картины, соответствующей расположению фиброзной бляшки (инфаркт миокарда, ишемический инсульт и т.д.), в связи с частичным или полным прекращением кровотока в поражённых сосудах.
ПАТОГЕНЕЗ АТЕРОСКЛЕРОЗА
Существуют три гипотезы, объясняющие возникновение атеросклероза: липидная, хронического повреждения эндотелия и моноклональная.
Липидная гипотеза. Предполагают, что остатки ЛП, богатых триглицеридами, захватываются макрофагами, что приводит к формированию ранних проявлений атеросклеротического процесса (стадия липидных полосок). Длительное пребывание ЛП в эндотелии сопровождается повреждением этих клеток, что, в свою очередь, приводит к отложению липидов во внеклеточном пространстве. Повреждение эндотелия и дальнейшее прогрессирование атеросклеротических изменений способствуют образованию фиброзной бляшки.
Повреждение эндотелия. Гипотеза хронического повреждения эндотелия базируется на том, что ряд таких факторов, как изменённый кровоток, увеличение концентрации холестерина ЛПНП, токсические и инфекционные агенты (вирусы, бактерии, хламидии), высокое содержание гомоцистеина могут приводить к повреждению поверхности внутренней оболочки артерии. Это ведёт к развитию хронического воспаления с во влечением макрофагов, Т-лимфоцитов, тромбоцитов и гладкомышечных клеток.
Моноклональная гипотеза (неопластическая гипотеза) основывается на предположении, что в основе атерогенеза находится мутация одного из многих генов, регулирующих клеточный цикл, что и приводит к пролиферации гладкомышечных клеток сосудистой стенки. Изменённые гладкомышечные клетки запускают атеросклеротический процесс.
ТИПЫ ГИПЕРЛИПИДЕМИЙ
Различают пять типов дислипидемий (гиперлипидемий, табл. 1-1).
• Тип I характеризуется очень высоким содержанием триглицеридов из-за увеличения концентрации хиломикронов.
• Тип Ia характеризуется высоким содержанием холестерина ЛПНП.
• Тип IIb отличает высокая концентрация триглицеридов и холестерина из-за увеличения содержания ЛПНП и ЛПОНП.
• Тип III возникает из-за накопления фрагментов хиломикронов и ЛППП. В сыворотке крови увеличено содержание холестерина и триглицеридов.
• Тип IV характеризуется увеличением содержания триглицеридов, нормальным уровнем холестерина и увеличением концентрации ЛПОНП.
• Тип V отличает увеличение преимущественно концентрации триглицеридов и холестерина.
Таблица 1-1. Типы гиперлипидемий по Фредриксону (1967)
Тип, относительная частота | Липид, вызывающий гипер липидемию | Лабораторные данные | Первичные гиперлипидемии | Вторичные гиперлипидемии |
Тип I, 1% | Преимущест-венно триглицериды | Гипер-хиломикронемия, недостаточность липопротеин липазы (ЛПЛазы) | Семейная недостаточность ЛПЛазы | Системная красная волчанка (СКВ), панкреатит, неадекватно контролируемый сахарный диабет |
Тип IIa, 10% | Холестерин | Увеличено содержание ЛПНП | Семейная гиперхолестеринемия | Гипотиреоз, нефроз, дисглобулинемия, острая порфирия, идио-патическая гиперкальциемия |
Тип IIb, 40% | Холестерин, триглицериды | Увеличено содержание ЛПНП и ЛПОНП | Семейная гиперхолестеринемия, семейная комбинированная гиперлипидемия | Нефротический синдром, сахарный диабет |
Тип III, 1% | Холестерин, триглицерид | Увеличено содержание ЛППП | Семейная дисбеталипопроте-инемия | Сахарный диабет, гипотиреоз, дисглобулинемия |
Тип IV, 45% | Триглицериды | Увеличено содержание ЛПОНП | Семейная гипертриглицеридемия, семейная комбинированная гиперлипидемия | Гликогенозы, гипотиреоз, СКВ, сахарный диабет, нефротический синдром, почечная недостаточность |
Тип V, 5% | Преимущест-венно триглицериды, холестерин | Увеличено содержание хиломикронов, ЛПОНП | Семейная гипертриглицеридемия, семейная комбинированная гиперлипидемия | Неадекватно контролируемый сахарный диабет, гликогенозы, гипотиреоз, нефротический синдром, дисглобулинемия, беременность, приём эстрогенов при семейной гипертри-глицеридемии |
Различают первичные (наследственные, генетические) и вторичные (при различных заболеваниях) дислипидемии.
Первичные гиперхолестеринемии
• Семейная гиперхолестеринемия (гиперлипидемия IIа по Фредриксону) — аутосомно-доминантное заболевание. В основе лежит дефект рецепторов ЛПНП. Гомозиготы встречаются с частотой 1 на 1 млн. Для них характерно увеличение содержания холестерина ЛПНП до 15-31 ммоль/л и развитие ИБС до 20-летнего возраста. Гетерозиготы (по сравнению с гомозиготами количество рецепторов ЛПНП уменьшено на 50%) встречаются с частотой 1 на 500. Содержание холестерина ЛПНП достигает 6-15 ммоль/л. ИБС у таких людей развивается между 30 и 40 годами.
• Полигенная гиперхолестеринемия возникает на фоне генетической предрасположенности под влиянием внешних факторов (ожирение, диета). Содержание в крови холестерина составляет 6-8 ммоль/л, ИБС развивается до 60 лет.
• Семейную комбинированную гиперлипидемию наблюдают у 1-2% населения (гиперлипидемия IIa, IIb, IV по Фредриксону).
Вторичные гиперхолестеринемии возникают при сахарном диабете, заболеваниях почек, патологии печени и желчевыводящих путей, гипотиреозе, панкреатитах, ожирении. Вторичная гиперлипидемия развивается значительно чаще первичной.
КЛИНИЧЕСКАЯ КАРТИНА
Длительное время (несколько десятилетий) атеросклеротический процесс протекает скрыто, и только потом появляются клинические признаки. Они зависят от преимущественной локализации процесса и степени обструкции сосудистого русла.
Поражаемые сосуды. Существуют типичные места преимущественной локализации атеросклеротического процесса: передняя межжелудочковая ветвь левой венечной артерии, бифуркация сонных артерий, проксимальные части почечных артерий.
• При поражении венечных артерий может возникнуть клиническая картина стенокардии, инфаркта миокарда или внезапная сердечная смерть.
• При поражении артерий мозга возникают транзиторные ишемические атаки или инсульт.
• Поражение атеросклеротическим процессом артерий нижних конечностей приводит к появлению перемежающейся хромоты (в том числе при синдроме Лериша) и гангрены.
• Атеросклеротический процесс в почечных артериях приводит к развитию стойкой артериальной гипертензии (АГ).
• При поражении брыжеечных артерий появляются симптомы ишемии кишечника.
Ксантомы, ксантелазмы, сенильная дуга. Внешними признаками атеросклеротического процесса могут быть ксантомы (бугристые образования в области суставов, пяточных сухожилий, обусловленные отложением холестерина), ксантелазмы (различной формы пятна на коже желтовато-оранжевого цвета, часто возвышающиеся, обусловленные отложением в коже холестерина и триглицеридов) и сенильная дуга на роговице (полоска желтоватого цвета по краю роговицы).
ДИАГНОСТИКА
Холестерин. Для диагностики нарушений липидного обмена обычно определяют содержание общего холестерина, холестерина ЛПВП, триглицеридов. По возможности проводят прямое определение концентрации холестерина ЛПНП (более дорогая и сложная методика). Чаще концентрацию холестерина ЛПНП подсчитывают по формуле Фридволда:
Холестерин ЛПНП (ммоль/л) =
Общий холестерин (ммоль/л) — Холестерин ЛПВП (ммоль/л) —
— 0,45 χ Триглицериды (ммоль/л)
где все значения приведены в ммоль/л.
Или:
Холестерин ЛПНП (мг%) =
Общий холестерин (мг%) — Холестерин ЛПВП (мг%) — 0,2 χ Триглицериды (мг%)
где все значения приведены в мг%.
Рекомендуемая оптимальная концентрация общего холестерина составляет не более 5 ммоль/л (190 мг%), а концентрация холестерина ЛПНП — не более 3 ммоль/л (115 мг%).
• Для оценки степени риска ИБС подсчитывают отношение общего холестерина к холестерину ЛПВП. Отношение более 5 указывает на высокий риск развития ИБС.
• Следует помнить, что концентрация триглицеридов плазмы может значительно изменяться в зависимости от приёма пищи и алкоголя. Концентрацию триглицеридов более 2 ммоль/л (180 мг%) считают показанием для её повторного определения.
• В острой стадии инфаркта миокарда и после операций на сердце концентрация общего холестерина, холестерина ЛПВП и холестерина ЛПНП может уменьшаться, а содержание триглицеридов увеличиваться.
Специальные методы диагностики — ангиография (см. главу 2 «Ишемическая болезнь сердца») и ультразвуковое исследование (УЗИ) сосудов — позволяют визуализировать фиброзные бляшки.
ЛЕЧЕНИЕ
Лечение дислипидемий начинают с немедикаментозных мер — изменения образа жизни, диеты (см. главу 3 «Профилактика ишемической болезни сердца», рис. 3-3). При отсутствии эффекта от немедикаментозных мероприятий назначают антигиперлипидемические средства.
Рис. 3-3. Алгоритм «Рекомендации по контролю за липидами» (Европейские рекомендации по профилактике сердечно-сосудистых заболеваний, 2003). ССЗ — сердечно-сосудистые заболевания.
Антигиперлипидемические средства
Применяют четыре основных класса антигиперлипидемических лекарственных средств (ЛС).
• Ингибиторы 3-гидрокси-3-метилглутарил-коэнзим А редуктазы (ГМГ-КоА редуктазы) — статины.
• Секвестранты жёлчных кислот — анионообменные смолы.
• Производные фибровой кислоты — фибраты.
• Препараты никотиновой кислоты.
Характеристика этих антигиперлипидемических средств приведена в табл. 1-2.
• Механизм эффекта статинов (ингибиторов ГМГ-КоА редуктазы) опосредован вмешательством в холестериновый каскад, протекающий в печени:
ацетат → ГМГ-КоА → мевалоновая кислота →
→ холестерин → жёлчные кислоты.
Конкурентное блокирование ГМГ-КоА редуктазы ведёт к уменьшению синтеза холестерина. Уменьшение синтеза холестерина вызывает по механизму обратной связи увеличение количества рецепторов ЛПНП в гепатоцитах, что приводит к захвату холестерина ЛПНП из плазмы крови и к снижению его уровня.
• Механизм действия секвестрантов жёлчных кислот (анионообменных смол) заключается в связывании жёлчных кислот в просвете кишечника и выделении их с фекалиями. Это стимулирует синтез в печени жёлчных кислот из холестерина. Уменьшение содержания эндогенного холестерина стимулирует его синтез, вызывает увеличение количества рецепторов ЛПНП в гепатоцитах и снижение концентрации холестерина ЛПНП в плазме. Таким образом, секвестранты более показаны при высокой концентрации холестерина ЛПНП и нормальной концентрации триглицеридов.
• Механизм действия фибратов (производные фибровой кислоты) заключается в увеличении активности липопротеин липаз (ЛПЛаз) и гидролизе триглицеридов, уменьшении синтеза ЛПОНП и увеличении распада ЛПНП.
• Основной механизм действия никотиновой кислоты заключается в торможении секреции печенью богатых триглицеридами ЛПНП и ЛПОНП (за счёт уменьшения мобилизации свободных жирных кислот из жировых депо). Никотиновая кислота особенно показана при высоком содержании в крови триглицеридов.
Сравнительная характеристика антигиперлипидемических средств. Существуют различия в воздействии антигиперлипидемических средств на липидный спектр. Статины, анионообменные смолы, никотиновая кислота эффективно снижают концентрацию холестерина ЛПНП, в то время как фибраты влияют на неё слабо. Холестерин ЛПВП слегка увеличивается при назначении статинов и смол, а при назначении никотиновой кислоты и фибратов его увеличение более значительно. Уровень триглицеридов умеренно уменьшается при назначении статинов и более выраженно — при лечении фибратами и никотиновой кислотой.
При отсутствии эффекта от монотерапии применяют комбинацию антигиперлипидемических средств. Её используют при смешанных гиперлипидемиях, в этом случае приём двух или более препаратов в меньших дозах даёт хороший эффект в случае непереносимости каждого в высоких дозах.
Преимущество в лечении дислипидемий следует отдавать ингибиторам ГМГ-КоА редуктазы (статинам) как классу липидснижающих препаратов, достоверно доказавших свою эффективность в уменьшении смертности вследствие ИБС, а также в увеличении продолжительности жизни. Возможна терапия как одним, так и несколькими препаратами.
Аферез холестерина ЛПНП
Удаление холестерина из крови путём фильтрации (аферез холестерина ЛПНП) рекомендовано при наличии выраженной гиперлипидемии — как правило, больным с семейной гомозиготной гиперхолестеринемией, а также пациентам с рефрактерной к лекарственному лечению гиперлипидемией.
Таблица 1-2. Антигиперлипидемические средства
Класс | Механизм действия | Эффект на ЛП | Названия, дозы (сутки) | Противопо- казания | Побочные эффекты | Рекомендации |
Статины | Подавляют активность ГМГ-КоА-редуктазы, уменьшают синтез холестерина, увеличивают синтез аполипопротеина А1 | ↓ЛПНП на 25-40%; ↓ЛПОНП | Ловастатин 10-80 мг (1); симвастатин 10-40 мг (1); правастатин 10-40 мг (1); флувастатин 20-40 мг (1); аторвастатин 10-80 мг (1) | Заболевания печени в острой стадии, беременность | Нарушения функций печени, миозит (редко) с повышением активности креатинфосфокиназы (КФК) в сыворотке крови, бессонница, расстройства желудочно-кишечного тракта (ЖКТ), аллергия | Принимать вечером, во время еды; первые 15 мес функциональные пробы печени каждые 3 мес; с осторожностью назначают на фоне алкоголя, больным с заболеваниями печени, артериальной гипотензией, эпилепсией, после черепно-мозговой травмы (ЧМТ) |
Фибраты | Увеличивают активность ЛПЛаз и гидролиз триглицеридов, уменьшают синтез ЛПОНП и увеличива-ют катаболизм ЛПНП | ↓триглице-риды на 25-40%; ↓ЛПОНП; ↓ЛПНП; -ЛПВП на 10-15% | Гемфиброзил 600 мг (2); фенофибрат 100 мг (3); безафибрат 200 мг (2-3); ципрофибрат 100 мг (1) | Беременность | Тошнота, нарушение функций печени, миозиты, анемия | Принимать за 30 мин до еды |
Никотиновая кислота | Подавляет синтез ЛПОНП и ЛПНП в печени, увеличивает активность ЛПЛаз | ↓триглицериды на 25-85%; ↓ЛПОНП на 25-35%; ↓ЛПНП на 15-25%; могут -ЛПВП | Никотиновая кислота 50-100 мг (2) (до 1-2 г/сут) | Нарушение функций печени, обострение пептической язвы, сахарный диабет, подагра | Эритема и зуд кожи, тахикардия, предсердные аритмии, расстройства ЖКТ, гиперурикемия, гипергликемия, нарушения функций печени | Рекомендуют употреблять продукты, богатые метионином; начинают с 500 мг/сут и постепенно увеличивают дозу до 3 г/сут в 1-3 приёма во время или после еды |
Источник