Нарушение синтеза гемоглобина порфирии
Содержание статьи
У гемоглобина есть молекулярные болезни
Серповидно-клеточная анемия
HbS – гемоглобин серповидно-клеточной анемии. При этом нарушении в ДНК в результате точковой мутации триплет ЦТТ заменен на триплет ЦАТ, что влечет за собой включение в 6-м положении β-цепи вместо глутамата аминокислоты валина. Изменение свойств β-цепи влечет изменение свойств всей молекулы и формирование на поверхности гемоглобина «липкого» участка. При дезоксигенации гемоглобина участок «раскрывается» и связывает одну молекулу дезоксигемоглобина S с другими подобными. Результатом является полимеризация гемоглобиновых молекул и образование крупных белковых тяжей, вызывающих деформацию эритроцитов и, при прохождении ими капилляров, гемолиз.
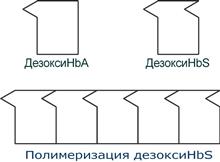
Схема отличия гемоглобина S от гемоглобина А и его полимеризация
Нарушение синтеза гемоглобина
Порфирии
Порфирии – это группа гетерогенных наследственных заболеваний, возникающих в результате нарушения синтеза гема и повышения содержания порфиринов и их предшественников в организме. Выделяют наследственные и приобретенные формы порфирии.
Приобретенные формы порфирий носят токсический характер и вызываются действием гексахлорбензола, солей свинца и других тяжелых металлов (ингибирование порфобилиногенсинтазы, феррохелатазы и др.), лекарственными препаратами (антигрибковый антибиотик гризеофульфин).
При наследственных формах дефект фермента имеется во всех клетках организма, но проявляется только в одном типе клеток. Можно выделить две большие группы порфирий:
1. Печеночные – группа заболеваний с аутосомно-доминантными нарушениями ферментов различных этапов синтеза протопорфирина IX.
Наиболее ярким заболеванием этой группы является перемежающаяся острая порфирия, при которой у гетерозигот активность уропорфириноген-I-синтазы снижена на 50%. В результате больные экскретируют с мочой большие количества порфобилиногена и аминолевулиновой кислоты. На свету порфобилиноген окисляется в окрашенные соединения и моча темнеет. Симптомами заболевания являются острые боли в животе, нервно-психические расстройства (полиневриты, тетрапарез, галлюцинации), сердечно-сосудистые нарушения.
Заболевание проявляется после достижения половой зрелости из-за повышенной потребности гепатоцитов в цитохроме Р450 для обезвреживания половых стероидов, обострение состояния также часто бывает после приема лекарственных препаратов, метаболизм которых требует участия цитохрома Р450. Снижение концентрации гема, используемого для синтеза цитохрома Р450, активирует аминолевулинат синтазу.
2. Эритропоэтические – аутосомно-рецессивные нарушения некоторых ферментов синтеза протопорфирина IX в эритроидных клетках. При этом при синтезе уропорфириногенов баланс реакций смещается в сторону синтеза уропорфириногена I. Примером может служить болезнь Гюнтера (частота<1:1000000), при которой развиваются высокая фоточувствительность кожи (ожоги под солнечным светом) и ее поражение, гемолиз, эритродонтия (окрашивание зубов в красный цвет), разрушение хрящей, в том числе носа и ушей.
Талассемии
Для талассемий характерно снижение синтеза α-цепей гемоглобина (α-талассемия) или β-цепей (β-талассемия). Это приводит к нарушению эритропоэза, гемолизу и тяжелым анемиям.
Источник
Порфирия (Порфириновая болезнь)
Порфирии ‒ большая группа наследственных заболеваний, характеризующихся нарушением биосинтеза гема и накоплением его токсичных метаболитов. Клинические проявления крайне разнообразны – от светочувствительности и кожных высыпаний до болей в животе, полного паралича и острых психозов. Диагностика осуществляется с помощью молекулярно-генетических тестов, специальных лабораторных методов определения порфиринов и их предшественников в моче и кале, оценки активности ферментов в крови. Лечение заключается в мероприятиях, направленных на снижение образования токсических метаболитов, их выведение из крови, проведении симптоматической терапии и хирургических вмешательств.
Общие сведения
Порфирии (от греч. «porphyreis» — пурпурный) – ряд заболеваний обмена веществ, при которых нарушается образование гема, в результате чего в организме накапливаются порфирины или их токсичные предшественники. Патологии данной группы встречаются относительно редко ‒ от 7 до 12 случаев на 100 000 человек. Отдельные нозологии имеют свою эндемичность. Так, распространенность поздней кожной порфирии в странах Южной Африки составляет 1:800, острой перемежающейся порфирии в Швеции ‒ 1:1000, вариегатной порфирии в Южной Африке ‒ 1:3000. У большинства порфирий нет гендерных различий, кроме поздней кожной формы (чаще страдают мужчины) и острой интермиттирующей (чаще болеют женщины).

Порфирия
Причины порфирий
В подавляющем большинстве случаев причиной порфирий выступают генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в биосинтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается вследствие заболеваний печени (алкогольный гепатит, вирусный гепатит С) или длительной интоксикации тяжелыми металлами. Наследование порфирий происходит по аутосомно-доминантному или аутосомно-рецессивному типу. Синтезирование гема протекает в 8 последовательных этапов, за каждый отвечает свой фермент, кодируемый определенным геном. Для каждой формы порфирии существует специфичный ферментативный дефект.
Гем представляет собой комплексное соединение порфиринов с двухвалентным железом. Наибольшее количество гема образуется в печени и костном мозге. В печени гем входит в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и обезвреживании различных ксенобиотиков. В костном мозге гем используется для образования гемоглобина. Результатом сниженной активности ферментов является торможение синтеза гема на определенном уровне, что ведет к накоплению его токсичных промежуточных метаболитов.
Помимо генетической мутации, для развития острых порфирий необходимо воздействие провоцирующих факторов, стимулирующих выработку порфиринов. Такими факторами являются голодание, длительная инсоляция, стрессы, алкоголь, инфекции, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства, подвергающиеся метаболизму системой цитохрома P-450 (нестероидные противовоспалительные препараты, антибиотики, антиконвульсанты, оральные контрацептивы, седативные средства). Особую роль играют колебания женских половых гормонов во время менструаций или беременности. У женщин месячные являются наиболее частым провоцирующим фактором, а беременность ассоциируется с тяжелым течением заболевания.
Патогенез
В результате неполноценности ферментов, участвующих в образовании гема, и действия провоцирующих факторов происходит увеличение концентрации его токсичных продуктов обмена. Для хронических порфирий характерно накопление протопорфирина, копропорфирина и упопорфирина. При острых формах возрастает количество порфобилиногена и дельта-аминолевулиновой кислоты (ДАЛК).
Порфирины накапливаются в коже и под действием ультрафиолетового излучения (солнечного света) запускают процесс перекисного окисления липидов, вызывая деструкцию и гибель клеток кожи. Копропорфирин и протопорфирин усиливают пигментацию кожи и ускоряют рост волос (гипертрихоз). Плохо растворимый в воде протопорфирин откладывается в клетках печени, закупоривает портальные тракты и желчные протоки. Отложение уропорфирина в эритроцитах приводит к их ускоренному разрушению в селезенке (гемолиз). Предшественники порфиринов (ДАЛК и порфобилиноген), накапливаясь в нервной ткани, вызывают демиелинизацию и аксональную дегенерацию нервных волокон.
Классификация
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность. Наиболее целесообразно выделять следующие виды порфирий:
- Эритропоэтические. К ним относятся врожденная эритропоэтическая порфирия (ВЭП, болезнь Гюнтера), эритропоэтическая протопорфирия (ЭПП). Основной клинический признак – поражение участков кожи, которые подвергаются воздействию прямых солнечных лучей (фотосенсибилизация). Данные патологии являются наиболее тяжелыми и имеют самый высокий процент летальности.
- Острые печеночные. Сюда включены острая перемежающаяся порфирия (ОПП), вариегатная порфирия (ВП), наследственная копропорфирия (НКП). Острые порфирии характеризуются приступообразным течением. Преимущественно страдает нервная система. При ВП и НКП также встречаются признаки фотосенсибилизации.
- Хронические печеночные. К ним относят позднюю кожную порфирию (ПКП), которая имеет наследственную и приобретенную формы. Это наиболее благоприятный вид порфирии.
Симптомы порфирий
Спектр клинических проявлений очень широк. Порфирии могут протекать в виде острых атак или хронически. Различия наблюдаются также в возрасте дебюта заболевания. Так, эритропоэтические порфирии манифестируют уже в дошкольном детстве (3-5 лет), острые порфирии — после полового созревания (14-16 лет), а спорадическая (приобретенная) форма ПКП — после 40 лет.
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
При поздней кожной форме возникает гиперпигментация участков кожи, подвергающихся постоянному воздействию солнечного света (лицо, шея, ушные раковины, верхняя часть груди, кисти рук). Кожа приобретает землистый или бронзовый оттенок. Также характерны гипертрихоз лобно-височной области лица, фотосенсибилизация, проявляющаяся повышенной ранимостью кожи и образованием пузырей с жидкостным содержимым. После вскрытия пузырей формируются эрозии. На местах разрешения эрозий образуются атрофические рубцы.
При эритропоэтических порфириях наблюдаются более выраженные признаки светочувствительности, чем при ПКП (ранимость, пузыри, эрозии). При длительном нахождении на свету появляется покраснение и сильное жжение кожи. Обширные эрозии оставляют после себя грубые рубцы на лице, что приводит к обезображиванию внешнего вида больного. В результате множественных рубцов на коже кистей рук развиваются контрактуры суставов, что значительно затрудняет их движения. Моча становится красной или розовой, а зубы окрашиваются в красно-коричневый цвет (эритродонтия). Из-за увеличенной селезенки могут появиться тяжесть или ноющие боли в левом подреберье. Специфический признак ЭПП – утолщение, огрубение и уплотнение кожи вокруг рта и глаз, на крыльях и спинке носа, на тыльных поверхностях кистей.
Осложнения
Нарушения порфиринового обмена ухудшают течение сердечно-сосудистых заболеваний, неблагоприятно влияют на углеводный метаболизм и повышают риск развития сахарного диабета 2 типа. Острые формы порфирий вследствие выраженной полинейропатии осложняются параличом дыхательной мускулатуры, аспирационной пневмонией, отеком головного мозга, тромбоэмболиями, рабдомиолизом. Постоянные эрозии кожных покровов могут привести к бактериальным инфекциям. При ЭПП из-за отложения нерастворимого в воде протопорфирина может развиться цирроз печени и печеночная недостаточность.
Диагностика
При подозрении на порфирию пациента направляют к врачу-гематологу. При постановке диагноза учитывается наличие заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, менструации, беременность). Лабораторная диагностика порфирий следующая:
- Клинико-биохимические анализы. При болезни Гюнтера в общем и биохимическом анализах крови выявляются признаки гемолиза (пойкилоцитоз, анизоцитоз, сфероцитоз, ретикулоцитоз, повышение непрямого билирубина и сывороточного железа), увеличение печеночных трансаминаз. У больных с острыми порфириями отмечается снижение уровня глюкозы и натрия, при ПКП — увеличение сывороточного железа и ферритина. Также у 80% с ПКП выявляются положительные маркеры вируса гепатита С.
- Специфические исследования. Для диагностики острых порфирий широко используется скрининговая проба Эрлиха (при смешивании специального реактива с мочой она окрашивается в красный цвет). Для ОПП характерно повышение ДАЛК и порфобилиногена в моче, для ВП — протопорфирина в кале, для НКП – копропорфирина в кале и моче. При ПКП в моче увеличено содержание уропорфирина, в кале – копропорфирина. При ЭПП наблюдается высокая концентрация протопорфирина в эритроцитах и кале, при ВЭП — уропорфирина в моче, кале и эритроцитах. При люминесцентной микроскопии плазма дает красное флуоресцирование при ПКП и эритропоэтических порфириях.
Также для подтверждения диагноза проводится определение уровня ферментов цикла биосинтеза гема в эритроцитах, лимфоцитах или плазме — порфобилиногендезаминазы (ОПП), копропорфириноген-оксидазы (НКП), протопорфириноген-оксидазы (ВП), уропорфириногенсинтетазы (ВЭП), уропорфириногендекарбоксилазы (ПКП), феррохелатазы (ЭПП). Заключительным этапом диагностики является молекулярно-генетическое тестирование для выявления мутаций генов, кодирующих перечисленные выше ферменты. Данные исследования особенно эффективны для распознавания асимптомных форм порфирий.
Эритропоэтические порфирии дифференцируют с дерматологическими заболеваниями (буллезным пемфигоидом, вульгарной пузырчаткой), с гематологическими патологиями, протекающими со спленомегалией (лейкозами, лимфомами, аутоиммунными гемолитическими анемиями) с болезнями почек. ПКП дифференцируют с заболеваниями печени, гемохроматозом, надпочечниковой недостаточностью. Острые порфирии следует дифференцировать с хирургическими заболеваниями, сопровождающимися сильной болью в животе, неврологическими и психиатрическими патологиями.
Лечение порфирий
Пациентов с острыми и эритропоэтическими порфириями необходимо госпитализировать отделение гематологии. Лечение ПКП возможно как в стационаре, так и в амбулаторных условиях. На сегодняшний день не существует эффективных методов, полностью ликвидирующих нарушения обмена порфиринов. Основной упор делается на патогенетическую и симптоматическую терапию, а также на устранение провоцирующих факторов. Способы лечения зависят от вида порфирий:
- Острые. Для подавления образования порфобилиногена и ДАЛК применяют гем-аргинат, производные АТФ (аденил, рибоксин) и большие дозы глюкозы с дальнейшим переходом на высокоуглеводную диету. Для купирования вегетативной симптоматики используют октреотид, для ускорения восстановления миелина в нервных волокнах — витамины группы В. При менструалозависимых атаках эффективна овариосупрессивная терапия. С этой целью назначают агонисты гонадотропин-рилизинг гормона.
- Эритропоэтические. Данные порфирии очень плохо поддаются терапии. Основное лечение – это защита кожных покровов от солнечного света (окна со стеклом, не пропускающим ультрафиолет, закрытая одежда, фотозащитные кремы, прием бета-каротина). Необходимо обрабатывать эрозии антисептическими растворами для профилактики инфекционных осложнений. При выраженном гемолизе показана спленэктомия. В некоторых случаях болезни Гюнтера эффективной оказывается трансплантация костного мозга. При ЭПП дополнительно назначаются гепатопротекторы (урсодезоксихолевая кислота, адеметионин) и антицирротическая терапия.
- ПКП. С целью удаления порфиринов и избытка железа проводят плазмаферез и флеботомию (кровопускания). При противопоказаниях к данным процедурам назначаются аминохинолиновые (хлорохин) и комплексообразующие (дефероксамин) препараты. Для уменьшения всасывания порфиринов в желудочно-кишечном тракте применяют энтеросорбенты (активированный уголь). Также используются солнцезащитные кремы. При наличии гепатита С необходима противовирусная терапия интерфероном-альфа и рибавирином.
Прогноз и профилактика
В большинстве случаев порфирии являются тяжелыми заболеваниями с неблагоприятным прогнозом. При эритропоэтических формах продолжительность жизни составляет около 30 лет, смерть наступает от интеркуррентных инфекций. ЭПП часто приводит к циррозу печени. При атаках острых порфирий летальный исход наблюдается в 15-20%, основная причина смерти – паралич дыхательных мышц. При ПКП прогноз благоприятный, тяжелых осложнений не происходит. Для предупреждения рецидивов рекомендуется избегать провоцирующих факторов – инфекций, голодания, стрессов, длительной инсоляции, употребления алкоголя и определенных лекарственных средств. Людям, у которых в семье есть больной порфирией, необходимо определять активность ферментов цикла синтеза гема и проводить ДНК-диагностику для выявления генетических мутаций.
Источник
Нарушения синтеза гема. Порфирии
Порфирии— гетерогенная группа
заболеваний, вызванная нарушениями
синтеза гема вследствие дефицита одного
или нескольких ферментов.
Классификации порфирий
Единой классификации порфирий нет.
Порфирии делят по причинам на:
Наследственные. Возникают при
дефекте гена фермента, участвующего в
синтезе гема;Приобретенные. Возникают при
ингибирующем влиянии токсических
соединений (гексохлорбензол, соли
тяжелых металлов — свинец) на ферменты
синтеза гема.
В зависимости от преимущественной
локализации дефицита фермента (в печени
или эритроцитах) порфирин делится на:
печеночные– наиболее распространенный
тип порфирина к нему относится острая
перемежающаяся порфирия (ОПП), поздняя
кожная порфирия, наследственная
копропорфирия, мозаичная порфирия;эритропоэтические– врожденная
эритропоэтическая порфирия (болезнь
Гюнтера), эритропоэтическая протопорфирия.
В зависимости от клинической картины,
порфирии делят на:
острые.
хронические.
Негативные последствия порфирий связаны
с дефицитом гема и накоплением в тканях
и крови промежуточных продуктов синтеза
гема – порфириногенов и продуктов их
окисления. При эритропоэтических
порфириях порфирины накапливаются в
нормобластах и эритроцитах, при печёночных
— в гепатоцитах.
Для каждого вида порфирии существует
определенный уровень ферментативного
дефекта, в результате накапливаются
продукты, синтезирующиеся выше этого
уровня. Эти продукты являются основными
диагностическими маркерами заболевания.
Порфириногены ядовиты, при тяжёлых
формах порфирий они вызывают
нейропсихические расстройства, нарушения
функций РЭС и повреждения кожи.
Нейропсихические расстройства при
порфириях связаны с тем, что аминолевулинат
и порфириногены являются нейротоксинами.
В коже на солнце порфириногены легко
превращаются в порфирины. Кислород при
взаимодействии с порфиринами переходит
в синглетное состояние. Синглетный
кислород стимулирует ПОЛ клеточных
мембран и разрушение клеток, поэтому
порфирии часто сопровождаются
фотосенсибилизацией и изъязвлением
открытых участков кожи.
Порфириногены не окрашены и не
флуоресцируют, а порфирины проявляют
интенсивную красную флуоресценцию в
ультрафиолетовых лучах. Избыток
порфиринов который выводиться с мочой,
придает ей темный цвет («порфирин» в
переводе с греч. означает пурпурный).
Иногда при лёгких формах наследственных
порфирии заболевание может протекать
бессимптомно, но приём лекарств,
являющихся индукторами синтеза
аминолевулинатсинтазы, может вызвать
обострение болезни. В некоторых случаях
симптомы болезни не проявляются до
периода полового созревания, когда
повышение образования β-стероидов
вызывает индукцию синтеза
аминолевулинатсинтазы. Порфирии
наблюдают и при отравлениях солями
свинца, так как свинец ингибирует
аминолевулинатдегидратазу и феррохелатазу.
Некоторые галогенсодержащие гербициды
и инсектициды являются индукторами
синтеза аминолевулинатсинтазы, поэтому
попадание их в организм сопровождается
симптомами порфирии.
Виды порфирий
Острая перемежающая порфирия(ОПП) – причина – дефект гена, кодирующего
ПБГ – дезаминазу. Наследуется по
аутосомно-доминатному типу. Происходит
накопление ранних предшественников
синтеза гема: 5- АЛК (5-ALA)
и порфобилиногена (ПБГ).
Бесцветный ПБГ на свету превращается
в порфибилин и порфирин, они предают
моче темный цвет. АЛК оказывает
нейротоксическое действие, приводя к
вялому параличу конечностей и парезу
дыхательной мускулатуры. Последнее
вызывает острую дыхательную недостаточность.
Заболевание проявляется в среднем
возрасте, провоцируется приемом
анальгетиков, сульфаниломидных
препаратов, так как они увеличивают
синтез АЛК – синтазы.
Клинической симптоматикой являются
острые боли в животе, рвота, запор,
сердечно-сосудистые нарушения,
нервно-психические расстройства. Не
наблюдается повышенной чувствительности
к свету, так как метаболическое нарушение
проходит на стадии, предшествующей
образованию уропорфириногена.
Для лечения применяют препарат нормосанг
– аргинат гема. Действие основано на
том, что гем, по механизму отрицательной
обратной связи блокирует трансляцию
АЛК – синтазы, а, следовательно, падает
синтез АЛК и ПБГ, чем и достигается
купирование симптоматики.
Врожденная эритропоэтическаяпорфирия -это еще
более редкое врожденное заболевание,
наследуемое по аутосомно-рецессивному
типу. Молекулярная природа этой
болезни точно неизвестна; установлено,
однако, что для нее характерен определенный
дисбаланс относительных активностей
уропорфириноген-Ш-косинтазы и
уропорфириноген-1-синтазы. Образование
уропорфириногена Iв
количественном отношении значительно
превосходит синтез уропорфириногенаIII—нормального изомера
на пути синтеза гема. Хотя генетическое
нарушение распространяется на все
клетки, проявляется оно по неизвестной
причине преимущественно в эритропоэтической
ткани. Пациенты с врожденной
эритропоэтической порфирией экскретируют
большие количества изомеров типа
Iуропорфириногена и копропорфириногена;
в моче оба этих соединения самопроизвольно
окисляются в уропорфирин Iи копропорфиринI—красные
флуоресцирующие пигменты. Сообщалось
о случае, когда наблюдалось небольшое
повышение концентрации уропорфиринаIII, но отношение изомеров
типа Iи IIIсоставляло примерно 100:1.Циркулирующие эритроциты содержат
большое количество уропорфирина
1,однако, наивысшая концентрация
этого порфирина отмечена в клетках
костного мозга (но не в гепатоцитах).
Отмечается светочувствительность
кожи,обусловленная характером спектра
поглощения порфириновых соединений,
которые образуются в больших
количествах. У пациентов отмечаются
трещины на коже, часто наблюдаются
гемолитические явления.
Наследственная копропорфирия
—аутосомно-доминантное нарушение,
обусловленное дефицитомкопропорфнрнногеноксидазы
—митохондриального фермента,
ответственного за превращение
копропорфириногена IIIв
протопорфириноген IX.Копропорфириноген IIIв
больших количествах удаляется из
организма в составе фекалий, а также
вследствие его растворимости в воде
экскретируется в большом количестве с
мочой. Как и уропорфириноген,
копропорфириноген на свету и воздухе
быстро окисляется, превращаясь в красный
пигмент копропорфирин.
Ограниченная
при этом заболевании способность к
синтезу гема (особенно в стрессовых
условиях) приводит к дерепрессии
АЛК-сиитазы. В результате наблюдается
избыточное образование АЛК и
порфобилиногена, а также других
интермедиатов на пути синтеза тема,
образующихся на стадиях, предшествующих
наследственно заблокированному этапу.
Соответственно у пациентов с наследственной
копропорфирией обнаруживаются все
признаки и симптомы, связанные с избытком
АЛК и порфобилиногена, которые характерны
для перемежающейся острой порфирии, но
помимо этого у них имеется повышенная
светочувствительность, обусловленная
присутствием избыточных количеств
копропорфириногенов и уропорфириногенов.
При этом заболевании введение гематина
также может вызвать по крайней мере
частичную репрессию АЛК-синтазы и
смягчение симптомов, обусловленных
перепроизводством интермедиатов
биосинтеза гема.
Мозаичная порфирия,или
наследственная фоторопорфирия, является
аутосомно-доминантным нарушением, при
котором происходит частичное блокирование
ферментативного превращения
протопорфириногена в гем. В норме это
превращение осуществляется двумя
ферментами, протопорфириногеноксидазой
и феррохелатазой, локализованными
в митохондриях.Судя по данным,
полученным на культуре фибробластов
кожи, у больных мозаичной порфирией
содержание протопорфириногеноксидазы
составляет лишь половину нормального
количества. У пациентов с мозаичной
порфирией наблюдается относительная
недостаточность содержания гема в
стрессовых условиях, а также
дерепрессированное состояние печеночной
АЛК-синтазы. Как отмечалось выше,
повышенная активность АЛК-синтазы ведет
к перепроизводству всех интермедиатов
синтеза гема на участках перед
заблокированной стадией. Таким образом,
пациенты с мозаичной порфирией
экскретируют с мочой избыточные
количества АЛК, порфобилиногена,
уропорфирина и копропорфирина, а с
фекалиями выделяют уропорфирин,
копропорфирин и протопорфирин. Моча
больных пигментирована и флуоресцирует,
а кожа чувствительна к свету так же, как
и у больных поздней кожной порфирией
(см. ниже).
Поздняя кожная порфирия,вероятно, является наиболее распространенной
формой порфирии. Обычно она связана с
теми или иными поражениями печени,
особенно при избыточном потреблении
алкоголя или перегрузке ионами
железа. Природа метаболического
нарушения точно не установлена, но
вероятной причиной является частичная
недостаточность уропорфириноген-декарбоксилазы.
Нарушение, по-видимому, передается
как аутосомно-доминантный признак, но
генетическая пенетрантность различна
и в большинстве случаев зависит от
наличия нарушений функций печени. В
соответствии с предсказаниями моча
содержит повышенные количества
уропорфиринов типа Iи
III;в то же время экскреция с мочой
АЛК и порфобилиногена наблюдается
сравнительно редко. Иногда моча содержит
весьма значительное количество
порфиринов, придающих ей розоватый
оттенок; при подкислении она чаще всего
дает в ультрафиолетовой области розовую
флуоресценцию.
Печень содержит большие количества
порфиринов и поэтому сильно
флуоресцирует, тогда как у эритроцитов
и клеток костного мозга флуоресценция
отсутствует. Главным клиническим
проявлением при поздней кожной
порфирии является повышенная
светочувствительность кожи.У больных
не наблюдается ни повышенной активности
АЛК-синтазы, ни соответственно избыточного
содержания в моче порфобилиногена
и АЛК; это коррелирует с отсутствием
острых приступов, характерных для
перемежающейся острой порфирии.
Протопорфирия,или
эритропоэтическая протопорфирия,
по-видимому, обусловлена доминантно
наследуемой недостаточной активностью
феррохелатазыв митохондриях всех
тканей; клинически эта болезнь проявляется
как острая крапивница, вызываемая
воздействием солнечных лучей. Эритроциты,
плазма и фекалии содержат повышенные
количества протопорфирина
IX,а ретикулоциты (незрелые эритроциты)
и кожа (при исследовании с помощью
биопсии) часто флуоресцируют красным
светом. Печень, вероятно, тоже вносит
вклад в повышение образования
протопорфирина IX,однако
экскреции с мочой порфиринов и их
предшественников не наблюдается.
Синтез гемоглобина
Синтезированный в митохондриях гем
индуцируется синтез цепей глобина на
полирибосомах. Гены цепей глобина
расположены в 11 и 16 хромосоме.
Цепи глобина формируют глобулы и
соединяются с гемом. 4 глобулы нековалентно
соединяются в гемоглобин.
Гемоглобин начинает синтезироваться
на стадии базофильного эритробласта,
а заканчивается у ретикулоцитов. В
ретикулоцитах также идет синтез пуринов,
пиримидинов, фосфатидов, липида.
Чувствительным биохимическим индикатором
для отличия ретикулоцитов от зрелых
клеток является утрата последними
глутаминазы. Глутамин в ретикулоцитах
— источник углерода для синтеза порфирина
и азота для синтеза пурина.
Строение гемоглобина
Гемоглобин— тетрамерный
хромопротеин, имеет массу 64500Да, состоит
из 4 гемов и 4 глобинов. Глобины представлены
полипептидными цепями различных типов,,,и т.д.-цепь содержит
141 АК, а- цепь – 146
АК. Отдельные участки полипептидных
цепей образуют правозакрученные-спирали, особое
расположение в пространстве которых
формирует глобулы. Глобула-субъединицы
содержит 8-спиралей,
а-субъединицы –7.
Гем располагается в щелях между Е иFспиралями глобина, прикрепляясь через
гистидинF8 к спиралиFс помощью 5 координационной
связи железа. Гидрофобные остатки
аминокислот окружающие гем, препятствуют
окислению железа водой. 4 глобулы с
участием гидрофобных, ионных и водородных
связей формируют шарообразный тетрамер
гемоглобина. Максимально прочные связи,
в основном за счет гидрофобных связей,
образуются между-
и-глобулами. В
результате образуются 2 димера11и22.
Димеры соединяются между собой в основном
полярными (ионными и водородными)
связями, поэтому взаимодействие димеров
зависит от рН. Димеры легко перемещаются
друг относительно друга. В центре
тетрамера глобулы прилегают друг к
другу неплотно, образуя полость.
Функции гемоглобина
Обеспечивают перенос кислорода от
легких к тканям. В сутки около 600 литров;Участвует в переносе углекислого газа
и протонов от тканей к легким;Регулирует КОС крови.
Источник