Причиной анемии у человека может быть мутация в гене гемоглобина
Содержание статьи
Гемоглобинопатия — аномальные варианты гемоглобина | Университетская клиника
Гемоглобинопатия – это наследственные заболевания с единой проблемой – образованием аномальной формы гемоглобина, например, серповидноклеточная анемия S и талассемия.
Гемоглобинопатии носят эндемический характер – они возникают в определенном географическом районе, например, в Средиземноморье, Африке, Юго–Восточной Азии. В нашей стране они тоже встречаются.
Что такое гемоглобинопатия
Гемоглобинопатии – это заболевания, вызванные выработкой и присутствием аномальной формы гемоглобина.
Гемоглобин состоит из гема (частей, содержащих железо) и глобина (частей белка, состоящих из аминокислотных цепей). Молекулы гемоглобина (Hb или Hgb) находятся в красных кровяных тельцах. Их задача – связывать кислород в легких и передавать его тканям и органам, где они его выделяют.
Строение гемоглобина
Существует несколько типов цепей глобина: альфа, бета, дельта и гамма.
Типы нормального гемоглобина:
- A – HbA: составляет около 95-98% от общего гемоглобина у взрослых людей. Он содержит 2 альфа (α) цепи и две бета (β) цепи.
- A2 – HbA2: составляет около 2-3% от общего гемоглобина. Он содержит 2 цепи альфа (α) и две цепи дельта (δ).
- F (HbF): составляет около 2% от общего гемоглобина взрослого человека. Он содержит 2 альфа (α) цепи и две гамма (γ) цепи. Этот гемоглобин в основном вырабатывается у плода, его производство значительно снижается вскоре после рождения и достигает уровня взрослого человека в течение 1-2 лет.
К гемоглобинопатиям относятся: структурные варианты гемоглобина, гемоглобин S, серповидноклеточная анемия, гемоглобинопатия C, гемоглобинопатия E, талассемия, гемоглобин Бартс, наследственная персистенция гемоглобина плода.
Причины развития гемоглобинопатии
Гемоглобинопатии возникают в случае генетических изменений генов глобина, которые приводят к изменению аминокислот, составляющих белок глобина. Эти изменения влияют на:
- структуру гемоглобина, например, гемоглобин S, который вызывает серповидно-клеточную анемию;
- его поведение;
- количество продуцируемого вещества (талассемия);
- стабильность.
Серповидно-клеточная анемия
Существует четыре гена, кодирующих цепь альфа-глобина, и два гена, кодирующих цепь бета-глобина. Наиболее частым заболеванием, связанным с изменением альфа-цепи, является альфа-талассемия. Его тяжесть зависит от количества пораженных генов.
Талассемия характеризуется снижением продукции одной из цепей глобина, дисбалансом между альфа- и бета-цепями в гемоглобине A (альфа-талассемия) или увеличением малых форм, таких как Hb A2 или Hb F (бета-талассемия).
Изменения бета-цепей гемоглобина являются врожденными, аутосомно-рецессивными. Это означает, что больной человек должен иметь две дефектные копии гена, каждая от одного из родителей. Если один ген нормален, а другой дефектен, человек гетерозиготен, и мы называем его носителем. Аномальный ген может быть передан любому из потомков. Если рассматриваемый человек является гетерозиготным носителем, он может не иметь никаких симптомов и носительство не влияет на его здоровье.
Если происходят две модификации одного и того же бета-гена, человек гомозиготен по этому гену. Его организм может производить дефектный гемоглобин – возникает гемоглобинопатия с симптомами и потенциальными осложнениями. Степень тяжести зависит от генетической мутации и варьируется от случая к случаю. Копию гена можно передать потомству.
Если два аномальных бета-гена являются врожденными, человек является двойным, смешанным гетерозиготным. У него будут симптомы одной или обеих гемоглобинопатий. Один из аномальных бета-генов будет передаваться каждому из потомков.
Были идентифицированы сотни гемоглобинопатий в бета-цепях. Хотя лишь некоторые из них являются общими и клинически значимыми.
Клинические признаки и симптомы
Признаки и симптомы различаются по типу гемоглобинопатии и возможному сочетанию нескольких гемоглобинопатий. Некоторые приводят к усилению распада эритроцитов (гемолизу), уменьшению их общего количества и развитию анемии.
Клинические признаки включают:
- слабость, утомляемость;
- недостаток энергии;
- желтуха;
- бледность кожи.
Утомляемость
К серьезным клиническим признакам относятся:
- приступы сильной боли;
- удушье;
- увеличение селезенки;
- нарушения роста у детей;
- боль в верхней части живота (вызванная желчными камнями).
Удушье
Общие гемоглобинопатии
Красные кровяные тельца, содержащие аномальный гемоглобин, могут не переносить кислород достаточно эффективно. Они могут разрушаться раньше (чем в здоровых клетках крови) и развиваться гемолитическая анемия. Выявлены сотни гемоглобинопатий, но лишь некоторые из них являются общими и клинически значимыми.
Одной из наиболее распространенных гемоглобинопатий является серповидно-клеточная анемия с присутствием гемоглобина S. Это приводит к изменению формы – серповидно-клеточной – эритроцитов и снижению их выживаемости. Гемоглобин С может вызвать легкую гемолитическую анемию. Гемоглобин E обычно не приводит к развитию каких-либо или только очень легких клинических симптомов.
- Талассемия: самая распространенная генетическая аномалия в мире. Она часто встречается в Средиземноморье, на Ближнем Востоке и в Юго-Восточной Азии. Более легкая форма талассемии также встречается, например, у людей, родившихся в Чехии.
- Гемоглобин S: это основной гемоглобин людей с серповидно-клеточной анемией. В среднем эта мутация есть в одном из двух бета-генов у 8% американцев и африканцев. Возникновение этих мутаций в наших широтах встречаеся редко. Пациенты с заболеванием HbS имеют две аномальные цепи бета (b s) и две нормальные цепи альфа (a). Когда эритроциты, содержащие гемоглобин S, подвергаются действию пониженного количества кислорода (как это может быть в случае повышенной физической нагрузки или инфекционного заболевания легких), они деформируются, принимая форму полумесяца. Серповидные эритроциты могут блокировать периферические кровеносные сосуды и вызывать нарушения кровотока и боль. У них пониженная способность переносить кислород и более короткий срок жизни. Одна копия б не вызывает клинических проявлений, если не сочетается с другой мутацией гемоглобина, такой как HbC (b C) или бета-талассемия.
- Гемоглобин C: около 25% жителей Западной Африки и 2-3% афроамериканцев гетерозиготны по гемоглобину C (у них есть одна копия B C). Но заболевают только гомозиготные люди с обоими дефектными генами (b C). Обычные симптомы – легкая гемолитическая анемия с небольшим или средним увеличением селезенки.
- Гемоглобин E: вторая по распространенности гемоглобинопатия в мире с изменением бета-цепей. Патология очень часто встречается в Юго-Восточной Азии, особенно в Камбодже, Лаосе и Таиланде, а также частично в Северо-Восточной Азии. Есть случаи и в нашей стране. Люди с гомозиготным Hb E (две копии b E) обычно имеют легкую гемолитическую анемию, микроциты (маленькие красные кровяные тельца) и слегка увеличенную селезенку. Одна копия гемоглобина E не вызывает клинических признаков, если не сочетается с другой мутацией, такой как одна из бета-талассемии.
Талассемия
Необычные гемоглобинопатии
Существует ряд гемоглобинопатий, некоторые из которых не проявляются – они не вызывают никаких клинических признаков или симптомов. Другие, в свою очередь, влияют на функциональность и / или стабильность молекулы гемоглобина. Примерами являются гемоглобин D, гемоглобин G, гемоглобин J, гемоглобин M и гемоглобин Constant Spring. Мутации в гене альфа-цепи глобина приводят к образованию аномально длинных альфа (а) цепей, которые вызывают нестабильность в молекуле гемоглобина.
Другие примеры мутаций бета-цепи:
- Гемоглобин F: Hb F в основном вырабатывается в организме будущего ребенка (плода), и его функция заключается в переносе кислорода в среде с низким содержанием кислорода. Продукция гемоглобина F снижается сразу после рождения и стабилизируется на уровне взрослого человека до 1-2 лет. Гемоглобин F может быть повышен при некоторых врожденных заболеваниях. При бета-талассемии его уровень может быть нормальным или повышенным, но часто повышен при серповидно-клеточной анемии и сочетании серповидно-клеточной анемии с бета-талассемией. Пациенты с серповидно-клеточной анемией и повышенным Hb F часто имеют более легкое течение болезни, поскольку Hb F предотвращает серповидное движение красных кровяных телец. Уровни Hb F повышены в редком состоянии, называемом врожденным постоянством выработки гемоглобина плода (HPFH). Люди с повышенным уровнем гемоглобина F не имеют клинических признаков. HPFH вызывается разными генными мутациями у разных этнических групп. Hb F также может быть повышен при некоторых приобретенных состояниях, влияющих на выработку красных кровяных телец. Например, лейкемия и миелопролиферативные заболевания часто сопровождаются небольшим повышением уровня гемоглобина F.
- Гемоглобин H: HbH – это аномальный гемоглобин, который возникает в некоторых случаях альфа-талассемии. Его образование является ответом на фундаментальный недостаток альфа (а) цепей. Hb H состоит из четырех цепей бета (b) глобина. Хотя каждая из цепей бета-глобина нормальна, комплекс из четырех цепей бета нормально не функционирует. Обладает повышенным сродством к кислороду, плохо выделяет кислород клеткам тканей. Присутствие гемоглобина H также связано со значительным распадом эритроцитов (гемолизом), который возникает в результате осаждения нестабильного гемоглобина внутри красных кровяных телец.
- Hemoglobin Barts: Hb Barts вырабатывается в организме будущего ребенка с альфа-талассемией при условии, что все четыре гена, отвечающие за производство гемоглобина альфа, отсутствуют. Таким образом, не может образовываться гемоглобин HbA, HbA 2 и HbF. Гемоглобин Бартс состоит из четырех гамма (g) цепей и имеет высокое сродство к кислороду. Это состояние несовместимо с жизнью и обычно приводит к внутриутробной гибели плода.
Некоторые люди могут унаследовать два гена с разными мутациями, каждый от одного из родителей. Таких людей называют двойными или смешанными гетерозиготами.
Обследование: лабораторные тесты
Исследование на гемоглобинопатию проводится в следующих случаях:
- Выявление гемоглобинопатий у бессимптомных родителей больных детей.
- Выявление гемоглобинопатий у пациента с необъяснимой анемией, микроцитозом и / или гипохромией. Анализ может быть выполнен как часть теста на анемию.
- Скрининг на гемоглобинопатии у новорожденных – только в США и некоторых регионах с повышенной заболеваемостью.
- Пренатальный скрининг проводится в некоторых регионах с высокой частотой гемоглобинопатий (особенно в Африке).
На результаты тестов на гемоглобинопатию может повлиять переливание крови. Поэтому после переливания крови, прежде чем сдать анализ, пациенту следует подождать несколько месяцев. Тем не менее пациентам с серповидно-клеточной анемией после переливания крови рекомендуется сдать анализ крови, чтобы увидеть, достаточно ли гемоглобина в крови, и снизить риск повреждения организма серповидными эритроцитами.
Обследование гемоглобинопатий основано на обнаружении и оценке «нормальности» эритроцитов и гемоглобина в эритроцитах, а также на исследовании конкретной мутации гена. Каждый из тестов является частью головоломки, предоставляющей важную информацию о том, какая гемоглобинопатия присутствует. Для проверки гемоглобинопатии используются следующие тесты:
- Анализ крови. Анализ крови дает быструю информацию о клетках, циркулирующих в крови. Помимо прочего, результаты анализа крови показывают, сколько красных кровяных телец (эритроцитов) содержится в крови, какого они размера и формы, а также сколько там гемоглобина. Размер эритроцита определяет средний объем эритроцитов (MCV). Обнаружение пониженного MCV (микроцитоз, наличие небольших эритроцитов) часто сначала указывает на возникновение талассемии. Если MCV низкий и дефицит железа исключен, пациенты могут быть носителями талассемии или гемоглобинопатии, которые также вызывают микроцитоз (например, HbE).
- Анализ ДНК. Этот анализ используется для скрининга мутаций и делеций в альфа- и бета-областях глобиновых генов. Иногда обследуются все члены семьи. Задача в том, чтобы определить конкретный тип мутации, встречающейся в семье, и выявить всех носителей. ДНК-тесты не являются обычным тестом, но они могут помочь диагностировать гемоглобинопатию и выявить носителей.
- Мазок периферической крови (микроскопический дифференциальный подсчет лейкоцитов, считываемый по мазку периферической крови). Тест проводится путем формирования тонкого слоя крови на предметном стекле и окрашивания его специальными красителями. Образец крови, обработанный таким образом, затем оценивается лаборантом под микроскопом. Специалист определяет количество и тип белых и красных кровяных телец и тромбоцитов. Оценивает, являются ли они нормальными и зрелыми.
Анализ крови
При гемоглобинопатии эритроциты могут быть в следующих формах:
- Микроциты (меньше нормального).
- Гипохромные (более бледные, с пониженным гемоглобином).
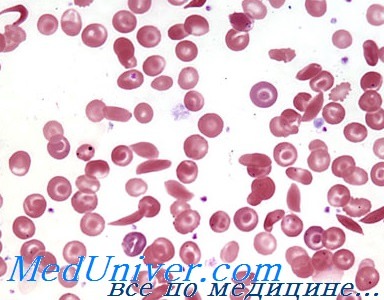
- Разных размеров (анизоцитоз) и формы (пойкилоцитоз, например, серповидно-клеточные клетки).
- С ядром (в незрелых эритроцитах) или с включениями.
- С неравномерным распределением гемоглобина (клетки-мишени, которые под микроскопом выглядят как «бычий глаз»).
Наличие более высокого процента аномально выглядящих эритроцитов означает более высокую вероятность наличия заболевания.
С помощью тестов на гемоглобинопатию и их комбинаций можно диагностировать наиболее распространенные гемоглобинопатии. Эти тесты могут помочь выявить пациентов с сочетанием различных гемоглобинопатий (смешанные гетерозиготы).
Лечение гемоглобинопатии
В настоящее время гемоглобинопатии – неизлечимые заболевания. Но возможно устранять симптомы заболевания. Цель – облегчить боль и минимизировать возможные осложнения. Также существуют лекарства, повышающие уровень гемоглобина F, что облегчает некоторые симптомы.
Однако исследования и поиск более безопасных и эффективных методов лечения все еще продолжается. В будущем для восстановления мутированного гена можно будет использовать трансплантацию стволовых клеток или генную терапию. Для того чтобы эти методы могли широко использоваться в будущем, необходимы дальнейшие обширные исследования.
Источники: БЕРТИС, Калифорния, ЭШВУД, Эр., Брунс, Делавэр, (ред.), Учебник Тиц по клинической химии и молекулярной диагностике. 4-е издание Луи: Эльзевье-Сондерс, 2006; LOTHAR, T. Клиническая лабораторная диагностика. Франкфурт: TH-Books, 1998; MASOPUST, J. Клиническая биохимия – требования и оценка биохимических исследований, часть I. и часть 2, Прага: Каролинум, 1998; RACEK, J., et al. Клиническая биохимия. 2. переработанное издание, Прага: Гален, 2006; Каспер Д.Л., Браунвальд Э., Фаучи А.С., Хаузер С.Л., Лонго Д.Л., редакторы Джеймсон Д.Л., 2005.
Поделиться ссылкой:
Источник
енетика серповидноклеточной анемии. Наследование
Генетика серповидноклеточной анемии. НаследованиеHbS был первым обнаруженным аномальным гемоглобином с высоким клиническим значением. Он возникает вследствие замены единственного нуклеотида, которая изменяет кодон шестой аминокислоты В-глобина глутаминовой кислоты на валин (GAG -> GTG: Glu6Val). Гомозиготность по данной мутации — причина серповидноклеточной анемии, серьезного заболевания, часто встречающегося в некоторых частях света. Болезнь имеет характерное географическое распределение, чаще всего встречается в Экваториальной Африке и реже всего в Средиземноморье, Индии и странах, в которые мигрировали люди из этих регионов. С этой, обычно фатальной в раннем детстве болезнью рождаются около 1 из 600 афроамериканцев, хотя все более частым становится более долгое выживание. Серповидноклеточная анемия — тяжелое аутосомно-рецессивное гемолитическое заболевание, характеризующееся тенденцией эритроцитов принимать выраженно аномальную форму (серпа) в условиях низкого насыщения кислородом. Гетерозиготы, про которых говорят, что они имеют «признак» серповидноклеточно-сти, обычно клинически здоровы, но их эритроциты в условиях очень низкого давления кислорода in vitro принимают форму серпа. Случаи, когда бы это могло происходить in vivo, редки, хотя гетерозиготы имеют риск инфаркта селезенки, особенно при полетах на большой высоте в самолетах с низким давлением в кабине. Гетерозиготное состояние наблюдают приблизительно у 8% афроамериканцев, но в областях, где частота гена высокая (например, в Западной Центральной Африке), вплоть до 25% новорожденных — гетерозиготы. Молекулярная патология HbS — серповидноклеточной анемииОколо 50 лет тому назад Ингрэм обнаружил, что аномалия HbS связана с заменой одной из 146 аминокислот в В-цепи молекулы гемоглобина. Все клинические проявления наличия HbS — последствия этого единственного изменения в гене В-глобина. Это было первой демонстрацией того, что мутация в структурном гене может вызывать замену аминокислоты в соответствующем белке. Поскольку аномалия HbS локализуется в В-цепи, формула HbS может быть записана как а2b2s или, более точно, а2Ab2s.
Гетерозиготы имеют смесь двух типов гемоглобинов (НbА и HbS), обозначаемых а2Аb2А, а2Аbs, а также гибридный тетрамер гемоглобина, обозначаемый как a2AbA,bs. Серповидноклеточность и ее последствияМолекулы гемоглобина, содержащие мутантные субъединицы b-глобина, нормальны по их способности выполнять их главную функцию связывания кислорода (если они не полимеризованы, как указано далее), но в ненасыщенной кислородом крови они растворимы в пять раз меньше по сравнению с нормальным гемоглобином. Относительная нерастворимость дезоксигемоглобина S является физической основой феномена серповидноклеточности. В условиях низкой кислородной напряженности молекулы HbS собираются в форме полимеров, формирующих стержни или волокна, искажающие форму эритроцитов. Эти уродливые эритроциты деформируются хуже, чем в норме, и, в отличие от нормальных красных кровяных клеток, не могут сжиматься, проходя через капилляры, тем самым блокируя ток крови и вызывая локальную ишемию. Происхождение мутаций гемоглобина SУ большинства лиц африканского происхождения нормальный ген b-глобина содержится в пределах фрагмента рестрикции размером в 7,6 килобазы ДНК. В то же время в определенных частях Африки, например в Гане и почти у 70% афроамериканцев, аллель серповидноклеточного глобина часто обнаруживают во фрагменте размером в 13 килобаз. Частая ассоциация серповидноклеточного глобина с 13-килобазовым фрагментом — поразительный пример неравновесного сцепления. В других частях Африки (например, в Кении) мутация серповидноклеточности обычно связана с фрагментом размером в 7,6 килобазы. Эти находки позволяют утверждать, что мутация серповидноклеточности возникла в Западной Африке в хромосоме, которая содержала ген р-глобина во фрагменте длиной 13 килобаз, и что подобная мутация, по крайней мере, один раз, независимо произошла где-то еще. Защита от малярии, обеспечиваемая данной мутацией у гетерозигот, обеспечила ее высокую частоту в областях, пораженных малярией. — Также рекомендуем «Генетика гемоглобинов HbС, Hammersmith. Наследование» Оглавление темы «Генетика гемоглобинопатий»:
|
Источник