Распад гемоглобина биохимия схема
Содержание статьи
Распад гема — многостадийный процесс
За сутки у человека распадается около 9 г гемопротеинов, в основном это гемоглобин эритроцитов.
Эритроциты в норме живут 90-120 дней, после чего лизируются в клетках ретикулоэндотелиальной системы – макрофагах селезенки (главным образом), купферовских клетках печени и макрофагах костного мозга. При разрушении эритроцитов в кровеносном русле высвобождаемый гемоглобин образует комплекс с белком-переносчиком гаптоглобином (фракция α2-глобулинов крови) и также переносится в клетки РЭС селезенки, печени и костного мозга.
Синтез билирубина
В клетках РЭС гем в составе гемоглобина окисляется молекулярным кислородом. В реакциях последовательно происходит разрыв метинового мостика между 1-м и 2-м пиррольными кольцами гема с их восстановлением, отщеплением железа и белковой части и образованием оранжевого пигмента билирубина. Высвобождаемое железо может либо запасаться в клетке в комплексе с ферритином, либо выделяться наружу и связываться с трансферрином.
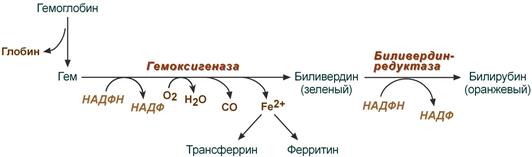
Реакции распада гемоглобина и образования билирубина
Билирубин – токсичное, жирорастворимое вещество, способное разобщать окислительное фосфорилирование в клетках. Особенно чувствительны к нему клетки нервной ткани.
Строение билирубина
Выведение билирубина
Из клеток ретикуло-эндотелиальной системы билирубин попадает в кровь. Здесь он находится в комплексе с альбумином плазмы, в гораздо меньшем количестве – в комплексах с металлами, аминокислотами, пептидами и другими малыми молекулами. Образование таких комплексов не позволяет выделяться билирубину с мочой. Билирубин в комплексе с альбумином называется свободный (неконъюгированный) или непрямой билирубин.
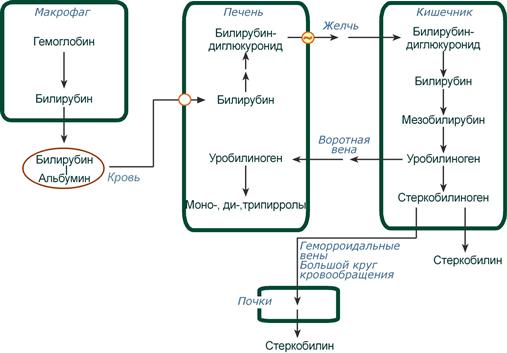
Этапы метаболизма билирубина в организме
Из сосудистого русла в гепатоциты билирубин попадает с помощью белка-переносчика (транспортный белок органических анионов) или по механизму флип-флоп. Далее при участии цитозольного связывающего белка лигандина (Y-протеин) билирубин транспортируется в ЭПР, где протекает реакция связывания билирубина с УДФ-глюкуроновой кислотой, при этом образуются моно- и диглюкурониды. Кроме глюкуроновой кислоты, в реакцию конъюгации могут вступать сульфаты, фосфаты, глюкозиды.
Билирубин-глюкуронид получил название связанный (конъюгированный) или прямой билирубин.
После образования билирубин-глюкурониды АТФ-зависимым переносчиком секретируются в желчные протоки и далее в кишечник, где при участии бактериальной β-глюкуронидазы превращаются в свободный билирубин. Одновременно, даже в норме (особенно у взрослых), некоторое количество билирубин-глюкуронидов может попадать из желчи в кровь по межклеточным щелям.
Таким образом, в плазме крови обычно присутствуют две формы билирубина: свободный (непрямой), попадающий сюда из клеток РЭС (80% и более всего количества), и связанный (прямой), попадающий из желчных протоков (в норме не более 20%).
Термины «связанный«, «конъюгированный«, «свободный«, «несвязанный» отражают взаимодействие билирубина и глюкуроновой кислоты (но не билирубина и альбумина!).
Термины «прямой» и «непрямой» введены, исходя из возможности химической реакции билирубина с диазореактивом Эрлиха. Связанный билирубин реагирует с реактивом напрямую, без добавления дополнительных реагентов, т.к. является водорастворимым. Несвязанный (жирорастворимый) билирубин требует добавочных реактивов, реагирует не прямо.
Превращение в кишечнике
В кишечнике билирубин подвергается восстановлению под действием микрофлоры до мезобилирубина и мезобилиногена (уробилиногена). Часть уробилиногена всасывается и с кровью портальной вены попадает в печень, где либо распадается до моно-, ди- и трипирролов, либо окисляется до билирубина и снова экскретируется. При этом при здоровой печени в общий круг кровообращения и в мочу мезобилирубин и уробилиноген не попадают, а полностью задерживаются гепатоцитами.
Оставшаяся в кишечнике часть пигментов ферментами бактериальной флоры толстого кишечника восстанавливается до стеркобилиногена. Далее
- малая часть стеркобилиногена может всасываться и катаболизировать в печени, подобно уробилиногену,
- незначительное количество стеркобилиногена через геморроидальные вены попадает в большой круг кровообращения, отсюда в почки и в мочу. После окисления на воздухе из стеркобилиногена образуется стеркобилин мочи,
- однако основное количество стеркобилиногена достигает нижних отделов толстого кишечника и выделяется. В прямой кишке и на воздухе стеркобилиноген окисляется в стеркобилин, окрашивая кал,
- аналогично уробилиноген, появляющийся в моче при патологии печени, окисляется в уробилин.
Очень часто стеркобилиноген, содержащийся в нормальной моче, называют уробилиногеном. И в клинической практике обычно не проводят различий между стеркобилиногеном и уробилиногеном мочи, их рассматривают как один пигмент – урохромы (уробилиноиды), что может создавать некоторую путаницу при оценке результатов анализа.
Источник
Биосинтез и распад гемоглобина — Студопедия
Тема 3 (вопрос 4 и 5).
Гем состоит из иона двухвалентного железа и порфирина. В основе структуры порфиринов находится порфин. Порфин представляет собой четыре пиррольных кольца, связанных между собой метеновыми мостиками. С наибольшей скоростью гем синтезируется в костном мозге (для синтеза гемоглобина ретикулоцитами) и в печени (для образования цитохрома Р-450).
Синтез гема происходит в несколько стадий:
1. На первой стадии в митохондриях сукцинил-КоА взаимодействует с глицином с образованием δ-аминолевулиновой кислоты. Эту реакцию катализирует специфический пиридоксальзависимый фермент δ-аминолевулинатсинтаза. Фермент активируется стероидами и ингибируется по типу обратной связи конечным продуктом — гемом. Продукт реакции из матрикса митохондрий переходит в цитозоль.
2. На второй стадии, в цитозоле, происходит конденсация 2-х молекул
δ-аминолевулиновой кислоты с образованием порфобилиногена. Фермент — порфобилиногенсинтаза — ингибируется конечным продуктом.
3. Из 4-х молекул порфобилиногена синтезируется протопорфирин IX, являющийся предшественником гема.
4. Протопорфирин IX присоединяет молекулу Fe при участии феррохелатазы (гемсинтаза) и образуется гем. Источником железа в этой реакции является белок ферритин, который депонирует железо.
Механизм регуляции синтеза тема в неэритроидных клетках имеет определенные отличия. Так, в клетках печени, где синтез гема происходит на высоком уровне, гем является отрицательным регулятором синтеза δ-аминолевулинатсинтазы по механизму репрессии-депрессии в процессе транскрипции. Главный регуляторный эффект гема состоит в том, что синтез фермента значительно ускоряется в отсутствии гема и замедляется в его присутствии.
Синтез белковой части гемоглобина происходит на рибосомах. Цепи α и β глобина синтезируются на полисомах, образованных, как правило, пятью рибосомами. Цепь α освобождается первой, присоединяется к β-цепи, еще связанной с рибосомой и отделяет ее, образуя димер αβ. Два димера соединяются в молекулу гемоглобина α2β2.
Соединение гема с глобином может происходить или в процессе синтеза полипептидных цепей, или после окончания синтеза глобина. Синтез полипептидных цепей происходит только в присутствии гема. При низкой концентрации гема синтез глобина замедляется. Отсюда следует, что синтез гема и глобина происходит координировано и ни один из этих компонентов не образуется в избыточном или недостаточном количестве.
Катаболизм гемоглобина
Эритроциты имеют короткое время жизни (примерно 120 дней). При физиологических условиях в организме взрослого человека разрушается около 1 — 2×1011 эритроцитов в сутки. Их катаболизм происходит, главным образом, в ретикулоэндотелиальных клетках селезёнки (РЭС), лимфатических узлов, костного мозга и печени. При распаде гемоглобина образуется билирубин. Билирубин является основным желчным пигментом у человека. При распаде 1 г гемоглобина образуется 35 мг билирубина, а в сутки у взрослого человека – примерно 250-350 мг. Дальнейший метаболизм билирубина происходит в печени.
Билирубин, образованный в клетках РЭС селезёнки и костного мозга, называется свободным (неконьюгированным)или непрямым, поскольку вследствие плохой растворимости в воде он легко адсорбируется на белках плазмы крови (альбуминах) и для его определения в крови необходимо предварительное осаждение белков спиртом. После этого билирубин определяют реакцией с диазореактивом Эрлиха. Свободный (непрямой) билирубин не проходит через почечный барьер и в мочу не попадает.
Каждая молекула альбумина связывает 2 (или 3) молекулы билирубина. При низком содержании альбумина в крови, а также при вытеснении билирубина из центров связывания на поверхности альбумина высокими концентрациями жирных кислот, лекарственных веществ (например, сульфаниламиды) увеличивается количество билирубина, не связанного с альбуминами. Он может проникать в клетки мозга и повреждать их.
Комплекс альбумин-билирубин с током крови попадает в печень, где происходит его превращение в прямой билирубин путем коньюгации с глюкуроновой кислотой. Реакцию катализирует УДФ-глюкуронилтрансфераза. Образующийся билирубиндиглюкуронид получил название прямого(коньюгированного) билирубина или связанного. Он растворим в воде и дает прямую реакцию с диазореактивом Эрлиха.
Прямой билирубин – это нормальный компонент желчи, попадающий в кровь в незначительном количестве. Он может проходить через почечный барьер, но в крови в норме его мало, поэтому в моче обычными лабораторными методами он не определяется.
Вместе с желчью прямой билирубин выводится в тонкий кишечник. В кишечнике билирубинглюкурониды гидролизуются специфическими бактериальными ферментами β-глюкуронидазами. Освободившийся билирубин под действием кишечной микрофлоры восстанавливается с образованием сначала мезобилирубина, а затем мезобилиногена(уробилиногена). Небольшая часть уробилиногенов, всасываясь в тонком кишечнике и верхнем отделе толстого, через систему воротной вены попадает в печень, где практически полностью разрушается до дипиррольных соединений. Уробилиноген при этом в общий кровоток не поступает и в моче не определяется.
Основная часть уробилиногена поступает в толстый кишечник, где под влиянием микрофлоры подвергается дальнейшему восстановлению с образованием стеркобилиногена. Образовавшийся стеркобилиноген почти полностью выделяется с калом. На воздухе он окисляется и превращается в стеркобилин, являющийся одним из пигментов кала. Небольшая часть стеркобилиногена попадает путем всасывания через слизистую толстого кишечника в систему нижней полой вены (через геморроидальные вены), доставляется в почки и выводится с мочой (4 мг/сутки).
Источник
Гемоглобинопатия — аномальные варианты гемоглобина | Университетская клиника
Гемоглобинопатия – это наследственные заболевания с единой проблемой – образованием аномальной формы гемоглобина, например, серповидноклеточная анемия S и талассемия.
Гемоглобинопатии носят эндемический характер – они возникают в определенном географическом районе, например, в Средиземноморье, Африке, Юго–Восточной Азии. В нашей стране они тоже встречаются.
Что такое гемоглобинопатия
Гемоглобинопатии – это заболевания, вызванные выработкой и присутствием аномальной формы гемоглобина.
Гемоглобин состоит из гема (частей, содержащих железо) и глобина (частей белка, состоящих из аминокислотных цепей). Молекулы гемоглобина (Hb или Hgb) находятся в красных кровяных тельцах. Их задача – связывать кислород в легких и передавать его тканям и органам, где они его выделяют.
Строение гемоглобина
Существует несколько типов цепей глобина: альфа, бета, дельта и гамма.
Типы нормального гемоглобина:
- A – HbA: составляет около 95-98% от общего гемоглобина у взрослых людей. Он содержит 2 альфа (α) цепи и две бета (β) цепи.
- A2 – HbA2: составляет около 2-3% от общего гемоглобина. Он содержит 2 цепи альфа (α) и две цепи дельта (δ).
- F (HbF): составляет около 2% от общего гемоглобина взрослого человека. Он содержит 2 альфа (α) цепи и две гамма (γ) цепи. Этот гемоглобин в основном вырабатывается у плода, его производство значительно снижается вскоре после рождения и достигает уровня взрослого человека в течение 1-2 лет.
К гемоглобинопатиям относятся: структурные варианты гемоглобина, гемоглобин S, серповидноклеточная анемия, гемоглобинопатия C, гемоглобинопатия E, талассемия, гемоглобин Бартс, наследственная персистенция гемоглобина плода.
Причины развития гемоглобинопатии
Гемоглобинопатии возникают в случае генетических изменений генов глобина, которые приводят к изменению аминокислот, составляющих белок глобина. Эти изменения влияют на:
- структуру гемоглобина, например, гемоглобин S, который вызывает серповидно-клеточную анемию;
- его поведение;
- количество продуцируемого вещества (талассемия);
- стабильность.
Серповидно-клеточная анемия
Существует четыре гена, кодирующих цепь альфа-глобина, и два гена, кодирующих цепь бета-глобина. Наиболее частым заболеванием, связанным с изменением альфа-цепи, является альфа-талассемия. Его тяжесть зависит от количества пораженных генов.
Талассемия характеризуется снижением продукции одной из цепей глобина, дисбалансом между альфа- и бета-цепями в гемоглобине A (альфа-талассемия) или увеличением малых форм, таких как Hb A2 или Hb F (бета-талассемия).
Изменения бета-цепей гемоглобина являются врожденными, аутосомно-рецессивными. Это означает, что больной человек должен иметь две дефектные копии гена, каждая от одного из родителей. Если один ген нормален, а другой дефектен, человек гетерозиготен, и мы называем его носителем. Аномальный ген может быть передан любому из потомков. Если рассматриваемый человек является гетерозиготным носителем, он может не иметь никаких симптомов и носительство не влияет на его здоровье.
Если происходят две модификации одного и того же бета-гена, человек гомозиготен по этому гену. Его организм может производить дефектный гемоглобин – возникает гемоглобинопатия с симптомами и потенциальными осложнениями. Степень тяжести зависит от генетической мутации и варьируется от случая к случаю. Копию гена можно передать потомству.
Если два аномальных бета-гена являются врожденными, человек является двойным, смешанным гетерозиготным. У него будут симптомы одной или обеих гемоглобинопатий. Один из аномальных бета-генов будет передаваться каждому из потомков.
Были идентифицированы сотни гемоглобинопатий в бета-цепях. Хотя лишь некоторые из них являются общими и клинически значимыми.
Клинические признаки и симптомы
Признаки и симптомы различаются по типу гемоглобинопатии и возможному сочетанию нескольких гемоглобинопатий. Некоторые приводят к усилению распада эритроцитов (гемолизу), уменьшению их общего количества и развитию анемии.
Клинические признаки включают:
- слабость, утомляемость;
- недостаток энергии;
- желтуха;
- бледность кожи.
Утомляемость
К серьезным клиническим признакам относятся:
- приступы сильной боли;
- удушье;
- увеличение селезенки;
- нарушения роста у детей;
- боль в верхней части живота (вызванная желчными камнями).
Удушье
Общие гемоглобинопатии
Красные кровяные тельца, содержащие аномальный гемоглобин, могут не переносить кислород достаточно эффективно. Они могут разрушаться раньше (чем в здоровых клетках крови) и развиваться гемолитическая анемия. Выявлены сотни гемоглобинопатий, но лишь некоторые из них являются общими и клинически значимыми.
Одной из наиболее распространенных гемоглобинопатий является серповидно-клеточная анемия с присутствием гемоглобина S. Это приводит к изменению формы – серповидно-клеточной – эритроцитов и снижению их выживаемости. Гемоглобин С может вызвать легкую гемолитическую анемию. Гемоглобин E обычно не приводит к развитию каких-либо или только очень легких клинических симптомов.
- Талассемия: самая распространенная генетическая аномалия в мире. Она часто встречается в Средиземноморье, на Ближнем Востоке и в Юго-Восточной Азии. Более легкая форма талассемии также встречается, например, у людей, родившихся в Чехии.
- Гемоглобин S: это основной гемоглобин людей с серповидно-клеточной анемией. В среднем эта мутация есть в одном из двух бета-генов у 8% американцев и африканцев. Возникновение этих мутаций в наших широтах встречаеся редко. Пациенты с заболеванием HbS имеют две аномальные цепи бета (b s) и две нормальные цепи альфа (a). Когда эритроциты, содержащие гемоглобин S, подвергаются действию пониженного количества кислорода (как это может быть в случае повышенной физической нагрузки или инфекционного заболевания легких), они деформируются, принимая форму полумесяца. Серповидные эритроциты могут блокировать периферические кровеносные сосуды и вызывать нарушения кровотока и боль. У них пониженная способность переносить кислород и более короткий срок жизни. Одна копия б не вызывает клинических проявлений, если не сочетается с другой мутацией гемоглобина, такой как HbC (b C) или бета-талассемия.
- Гемоглобин C: около 25% жителей Западной Африки и 2-3% афроамериканцев гетерозиготны по гемоглобину C (у них есть одна копия B C). Но заболевают только гомозиготные люди с обоими дефектными генами (b C). Обычные симптомы – легкая гемолитическая анемия с небольшим или средним увеличением селезенки.
- Гемоглобин E: вторая по распространенности гемоглобинопатия в мире с изменением бета-цепей. Патология очень часто встречается в Юго-Восточной Азии, особенно в Камбодже, Лаосе и Таиланде, а также частично в Северо-Восточной Азии. Есть случаи и в нашей стране. Люди с гомозиготным Hb E (две копии b E) обычно имеют легкую гемолитическую анемию, микроциты (маленькие красные кровяные тельца) и слегка увеличенную селезенку. Одна копия гемоглобина E не вызывает клинических признаков, если не сочетается с другой мутацией, такой как одна из бета-талассемии.
Талассемия
Необычные гемоглобинопатии
Существует ряд гемоглобинопатий, некоторые из которых не проявляются – они не вызывают никаких клинических признаков или симптомов. Другие, в свою очередь, влияют на функциональность и / или стабильность молекулы гемоглобина. Примерами являются гемоглобин D, гемоглобин G, гемоглобин J, гемоглобин M и гемоглобин Constant Spring. Мутации в гене альфа-цепи глобина приводят к образованию аномально длинных альфа (а) цепей, которые вызывают нестабильность в молекуле гемоглобина.
Другие примеры мутаций бета-цепи:
- Гемоглобин F: Hb F в основном вырабатывается в организме будущего ребенка (плода), и его функция заключается в переносе кислорода в среде с низким содержанием кислорода. Продукция гемоглобина F снижается сразу после рождения и стабилизируется на уровне взрослого человека до 1-2 лет. Гемоглобин F может быть повышен при некоторых врожденных заболеваниях. При бета-талассемии его уровень может быть нормальным или повышенным, но часто повышен при серповидно-клеточной анемии и сочетании серповидно-клеточной анемии с бета-талассемией. Пациенты с серповидно-клеточной анемией и повышенным Hb F часто имеют более легкое течение болезни, поскольку Hb F предотвращает серповидное движение красных кровяных телец. Уровни Hb F повышены в редком состоянии, называемом врожденным постоянством выработки гемоглобина плода (HPFH). Люди с повышенным уровнем гемоглобина F не имеют клинических признаков. HPFH вызывается разными генными мутациями у разных этнических групп. Hb F также может быть повышен при некоторых приобретенных состояниях, влияющих на выработку красных кровяных телец. Например, лейкемия и миелопролиферативные заболевания часто сопровождаются небольшим повышением уровня гемоглобина F.
- Гемоглобин H: HbH – это аномальный гемоглобин, который возникает в некоторых случаях альфа-талассемии. Его образование является ответом на фундаментальный недостаток альфа (а) цепей. Hb H состоит из четырех цепей бета (b) глобина. Хотя каждая из цепей бета-глобина нормальна, комплекс из четырех цепей бета нормально не функционирует. Обладает повышенным сродством к кислороду, плохо выделяет кислород клеткам тканей. Присутствие гемоглобина H также связано со значительным распадом эритроцитов (гемолизом), который возникает в результате осаждения нестабильного гемоглобина внутри красных кровяных телец.
- Hemoglobin Barts: Hb Barts вырабатывается в организме будущего ребенка с альфа-талассемией при условии, что все четыре гена, отвечающие за производство гемоглобина альфа, отсутствуют. Таким образом, не может образовываться гемоглобин HbA, HbA 2 и HbF. Гемоглобин Бартс состоит из четырех гамма (g) цепей и имеет высокое сродство к кислороду. Это состояние несовместимо с жизнью и обычно приводит к внутриутробной гибели плода.
Некоторые люди могут унаследовать два гена с разными мутациями, каждый от одного из родителей. Таких людей называют двойными или смешанными гетерозиготами.
Обследование: лабораторные тесты
Исследование на гемоглобинопатию проводится в следующих случаях:
- Выявление гемоглобинопатий у бессимптомных родителей больных детей.
- Выявление гемоглобинопатий у пациента с необъяснимой анемией, микроцитозом и / или гипохромией. Анализ может быть выполнен как часть теста на анемию.
- Скрининг на гемоглобинопатии у новорожденных – только в США и некоторых регионах с повышенной заболеваемостью.
- Пренатальный скрининг проводится в некоторых регионах с высокой частотой гемоглобинопатий (особенно в Африке).
На результаты тестов на гемоглобинопатию может повлиять переливание крови. Поэтому после переливания крови, прежде чем сдать анализ, пациенту следует подождать несколько месяцев. Тем не менее пациентам с серповидно-клеточной анемией после переливания крови рекомендуется сдать анализ крови, чтобы увидеть, достаточно ли гемоглобина в крови, и снизить риск повреждения организма серповидными эритроцитами.
Обследование гемоглобинопатий основано на обнаружении и оценке «нормальности» эритроцитов и гемоглобина в эритроцитах, а также на исследовании конкретной мутации гена. Каждый из тестов является частью головоломки, предоставляющей важную информацию о том, какая гемоглобинопатия присутствует. Для проверки гемоглобинопатии используются следующие тесты:
- Анализ крови. Анализ крови дает быструю информацию о клетках, циркулирующих в крови. Помимо прочего, результаты анализа крови показывают, сколько красных кровяных телец (эритроцитов) содержится в крови, какого они размера и формы, а также сколько там гемоглобина. Размер эритроцита определяет средний объем эритроцитов (MCV). Обнаружение пониженного MCV (микроцитоз, наличие небольших эритроцитов) часто сначала указывает на возникновение талассемии. Если MCV низкий и дефицит железа исключен, пациенты могут быть носителями талассемии или гемоглобинопатии, которые также вызывают микроцитоз (например, HbE).
- Анализ ДНК. Этот анализ используется для скрининга мутаций и делеций в альфа- и бета-областях глобиновых генов. Иногда обследуются все члены семьи. Задача в том, чтобы определить конкретный тип мутации, встречающейся в семье, и выявить всех носителей. ДНК-тесты не являются обычным тестом, но они могут помочь диагностировать гемоглобинопатию и выявить носителей.
- Мазок периферической крови (микроскопический дифференциальный подсчет лейкоцитов, считываемый по мазку периферической крови). Тест проводится путем формирования тонкого слоя крови на предметном стекле и окрашивания его специальными красителями. Образец крови, обработанный таким образом, затем оценивается лаборантом под микроскопом. Специалист определяет количество и тип белых и красных кровяных телец и тромбоцитов. Оценивает, являются ли они нормальными и зрелыми.
Анализ крови
При гемоглобинопатии эритроциты могут быть в следующих формах:
- Микроциты (меньше нормального).
- Гипохромные (более бледные, с пониженным гемоглобином).
- Разных размеров (анизоцитоз) и формы (пойкилоцитоз, например, серповидно-клеточные клетки).
- С ядром (в незрелых эритроцитах) или с включениями.
- С неравномерным распределением гемоглобина (клетки-мишени, которые под микроскопом выглядят как «бычий глаз»).
Наличие более высокого процента аномально выглядящих эритроцитов означает более высокую вероятность наличия заболевания.
С помощью тестов на гемоглобинопатию и их комбинаций можно диагностировать наиболее распространенные гемоглобинопатии. Эти тесты могут помочь выявить пациентов с сочетанием различных гемоглобинопатий (смешанные гетерозиготы).
Лечение гемоглобинопатии
В настоящее время гемоглобинопатии – неизлечимые заболевания. Но возможно устранять симптомы заболевания. Цель – облегчить боль и минимизировать возможные осложнения. Также существуют лекарства, повышающие уровень гемоглобина F, что облегчает некоторые симптомы.
Однако исследования и поиск более безопасных и эффективных методов лечения все еще продолжается. В будущем для восстановления мутированного гена можно будет использовать трансплантацию стволовых клеток или генную терапию. Для того чтобы эти методы могли широко использоваться в будущем, необходимы дальнейшие обширные исследования.
Источники: БЕРТИС, Калифорния, ЭШВУД, Эр., Брунс, Делавэр, (ред.), Учебник Тиц по клинической химии и молекулярной диагностике. 4-е издание Луи: Эльзевье-Сондерс, 2006; LOTHAR, T. Клиническая лабораторная диагностика. Франкфурт: TH-Books, 1998; MASOPUST, J. Клиническая биохимия – требования и оценка биохимических исследований, часть I. и часть 2, Прага: Каролинум, 1998; RACEK, J., et al. Клиническая биохимия. 2. переработанное издание, Прага: Гален, 2006; Каспер Д.Л., Браунвальд Э., Фаучи А.С., Хаузер С.Л., Лонго Д.Л., редакторы Джеймсон Д.Л., 2005.
Поделиться ссылкой:
Источник