Участок гена кодирующий одну из полипептидных цепей гемоглобина
Содержание статьи
ены гемоглобина человека
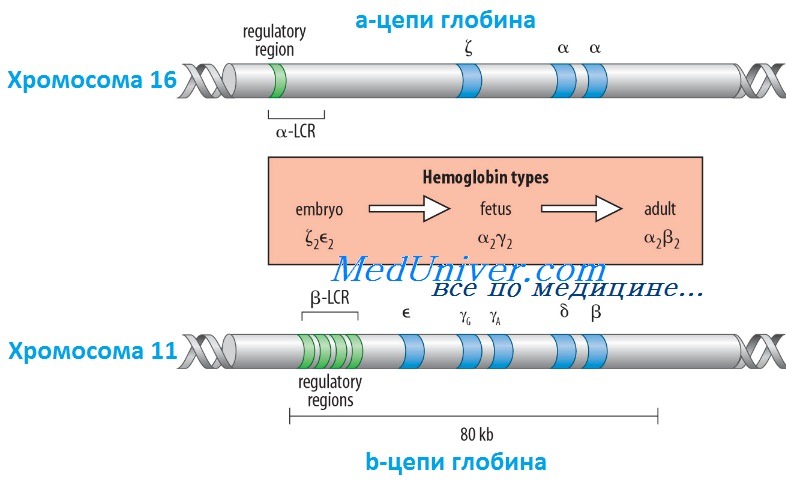
Гены гемоглобина человекаКроме НbА, у человека есть еще пять других нормальных гемоглобинов, которые имеют тетрамерные структуры, сравнимые с НbА и состоящие из двух а- или а-подобных цепей и двух не-b-цепей. Гены а- и а-подобных цепей расположены тандемно в хромосоме 16, а для b- и b-подобных — в хромосоме 11. В каждой копии хромосомы 16 есть два идентичных гена а-глобина, названные а1 и а2. В пределах комплекса генов b-глобина существует тесная гомология между разными генами. Например, b- и q-глобины отличаются только 10 из 146 аминокислот. Все гены глобина, несомненно, возникли из общего гена-предшественнника. Изменение экспрессии различных генов глобина в ходе развития иногда называют переключением глобинов. Это классический пример упорядоченного регулирования экспрессии генов в ходе развития. Гены в а- и b-группах размещаются в одной и той же транскрипционной ориентации и, что замечательно, гены внутри каждой группы расположены в той же последовательности, в которой они экспрессируются в процессе развития. Существует эквимолярное соответствие синтеза а- и b-подобных цепей глобина. Интересно, что переключение синтеза глобинов по времени сопровождается изменениями в основном месте эритропоэза. Эмбриональный синтез глобина происходит в желточном мешке с 3 по 8 нед гестации, но приблизительно около 5 нед гестации основное место кроветворения начинает перемещаться из желточного мешка в печень плода. HbF (а2у2) — преобладающий гемоглобин в внутриутробном периоде — составляет приблизительно 70% общего гемоглобина при рождении, но во взрослой жизни HbF составляет менее 1% общего гемоглобина. Хотя b-цепи могут обнаруживаться на ранних сроках гестации, их синтез становится значимым только ближе к сроку родов; к 3-месячному возрасту почти весь гемоглобин становится гемоглобином взрослого типа — HbА. Синтез 8-цепи также продолжается после рождения, но НbА2 (а2q2) никогда не составляет более примерно 2% гемоглобина взрослых. К несчастью, небольших количеств q-глобина (и, следовательно, HbA2) и у-глобина (и, следовательно, HbF), обнаруживаемых в норме в крови взрослого человека, недостаточно для компенсации сниженного количества b-глобина (и, следовательно, НbА), образующегося при болезнях типа b-талассемии. Следовательно, знание механизмов, регулирующих производство цепей глобина, потенциально имеет терапевтическое значение. Обнаружено множество факторов транскрипции, управляющих экспрессией генов глобина, что дает надежду на разработку лечения, направленного на увеличение синтеза q- и у-глобинов.
Регулирование экспрессии генов b-глобина: управляющий регион локусаКак и во многих других областях медицинской генетики, изучение механизмов, регулирующих экспрессию генов глобина, дало понимание как нормальных, так и патологических биологических процессов. Экспрессия гена b-глобина, как оказалось, только частично контролируется промотором и двумя энхансерами в фланговой ДНК, расположенной непосредственно рядом с геном. Необходимость дополнительных регулятор-ных элементов была заподозрена после идентификации уникальной группы пациентов, не имевших экспрессии ни одного гена в группе b-глобина, даже если сами гены (включая их индивидуальные регуляторные элементы) были неповрежденными. Оказалось, что такие пациенты имеют большие делеции выше комплекса b-глобина, удаляющие область приблизительно в 20 килобаз, названную локус-контролирующей областью (LCR, от англ. locus control region), которая начинается приблизительно в 6 килобазах выше гена е-глобина. Развивающаяся при этом болезнь, еу5р-талассемия, описана ниже. Данные пациенты показали, что LCR необходим для экспрессии всех генов в группе b-глобина в хромосоме 11. LCR определяется пятью сверхчувствительными к ДНКазе 1 участками, необходимыми для поддержки открытой конфигурации хроматина в данном локусе, что обеспечивает доступ факторам транскрипции к элементам, регулирующим экспрессию каждого гена в комплексе b-глобина. LCR вместе с ассоциированными связанными с ДНК белками, взаимодействует с генами локуса, формируя часть ядра, названную «транскрипционным узлом» (англ. active chromatin hub), в котором происходит экспрессия генов b-глобина. Последовательное переключение экспрессии генов, происходящее между пятью участниками комплекса гена b-глобина в ходе развития, вызвано последовательной ассоциацией транскрипционного узла с разными генами в группе, так как узел перемещается от 5′-конца комплекса (от экспрессирующегося в эмбриональном периоде гена e-глобина) через ген q до гена b-глобина у взрослых. Клиническое значение LCR разнообразно. Во-первых, пациенты с делециями LCR не экспрессируют гены группы b-глобина. Во-вторых, компоненты LCR, вероятно, окажутся существенными для генотерапии болезней группы b-глобина. В-третьих, знание молекулярных механизмов, лежащих в основе переключения глобинов, может сделать выполнимой, например, регуляцию экспрессии гена у-глобина у пациентов с b-талассемией (с мутациями в гене р-глобина), для стимуляции синтеза HbF (a2y2), — эффективного переносчика кислорода у взрослых с дефицитом НbА (а2b2). Для понимания патогенеза большинства гемоглобинопатий важны различия в дозе генов (четыре гена а-глобина и два гена b-глобина на диплоидный геном) и онтогенез а- и b-глобинов. Мутации в генах b-глобина более вероятно вызывают болезнь, чем мутации а-цепи, поскольку мутация единственного гена b-глобина влияет на 50% р-цепей, тогда как мутация одного гена а-цепи влияет только на 25% a-цепей. С другой стороны, мутации в гене b-глобина не имеют последствий во внутриутробном периоде, поскольку у-глобин является основным глобином до рождения, и к моменту родов HbF составляет три четверти общего гемоглобина. Поскольку а-цепи — единственный а-подобный компонент всех гемоглобинов, начиная с 6 нед после зачатия, мутации а-глобина вызывают тяжелую патологию как плода, так и послеродовой жизни. — Вернуться в содержание раздела «генетика» на нашем сайте Оглавление темы «Выявление генов болезни»:
|
Источник
ГЕМОГЛОБИНОПАТИИ
Гемоглобинопатии (haemoglobinopathia, ед. ч.; гемоглобин + греч. pathos страдание, болезнь; син. гемоглобиноз) — группа наследственных заболеваний, обусловленных наличием в эритроцитах аномальных гемоглобинов либо угнетением синтеза полипептидных цепей нормальных гемоглобинов. К Гемоглобинопатиям причисляют как выраженные патологические состояния, протекающие чаще с гемолитической анемией (см.), так и многочисленные случаи латентного носительства аномальных гемоглобинов или генов талассемии (см.).
После работ Л. Полинга, Итано (H. A. Itano) и сотр. (1949), посвященных гемоглобину S, и открытий в области биохимической генетики Гемоглобинопатии стали рассматривать как разновидность молекулярной патологии. В 1950 г. Итано и Дж. Нил описали аномальный гемоглобин С, в 1951 г. Итано описал аномальный гемоглобин D. В 1954 г. гемоглобин E одновременно, но независимо друг от друга описали Чернов, Минних и Чонгчареонсук (А. I. Chernoff, V. Minnich, S. Chongchareonsuk) и Итано, Берджер, Стерджен (W. R. Berger, P. Sturgeon). Гемоглобин G описали Эдингтон (G. М. Edington) и Г. Леманн в 1954 г. Известны также аномальные гемоглобины H, I, J, К, L, M, N, О, P, Q и др.
В мире насчитывают ок. 100 млн. человек — носителей аномальных гемоглобинов, однако эти цифры следует, по-видимому, считать заниженными вследствие несовершенства методов выявления аномальных гемоглобинов и недостаточной информации о распространении Гемоглобинопатий в различных областях земного шара.
Географическое распространение
В распространении Гемоглобинопатий имеют значение такие факторы, как принадлежность к определенным расовым и этническим группам, частота браков между кровными родственниками, миграция населения, заболеваемость тропической малярией. Вариации в синтезе гемоглобина встречаются преимущественно у жителей стран Южной Европы (побережье Средиземного моря), Африки и Азии или у выходцев из этих стран, среди негритянского населения Северной и Южной Америки. Более распространены Г. в зоне так наз. малярийного пояса земного шара, т. е. тропической, и несколько меньше — в субтропической зоне. Встречаются они и в некоторых южных республиках СССР (Азербайджан, Таджикистан).
Серповидная аномалия эритроцитов наиболее распространена в тропической Африке, странах Средиземноморья, Ближнего и Среднего Востока, Индии. Полагают, что только в тропической Африке насчитывают до 40 млн. человек — носителей аномального гемоглобина S. Не менее частой является, по-видимому, и талассемия. Носительство аномальных гемоглобином С и E более локализовано. Гетерозиготных носителей аномального гемоглобина С — более 7 млн. в Западной Африке и ок. 0,5 млн. среди негров США. В Юго-Восточной Азии (Индокитай, Таиланд, Бирма, Индонезия, Индия, Бангладеш и юж. районы Китая) носителей аномального гемоглобина E более 30 млн. человек. Основной очаг аномального гемоглобина D находится в Северо-Западной Индии, где количество гетерозиготных носителей определяется в несколько миллионов человек. Единичные случаи описаны в Северной и Западной Африке, Малой Азии и среди негритянского населения США. Очаги (бета-талассемии и спорадические случаи гемоглобинопатий S, С, D, E и альфа-талассемии обнаружены среди населения Азербайджана, Таджикистана и некоторых районов Кавказа.
Этиология и патогенез
Аномалия гемоглобина может возникать либо в результате качественных изменений его первичной структуры и функции, либо количественного уменьшения синтеза нормальных цепей глобина (см. Гемоглобин, генетика гемоглобина). Известно свыше 250 аномальных гемоглобинов; теоретически в молекуле гемоглобина возможно более 4000 различных точковых мутаций [Бил (D. Beale), Г. Леманн, 1965], из которых с помощью современных методов могут быть дифференцированы с нормальным гемоглобином только ок. 1500. В это число включены мутации типа чистых аминокислотных замещений, при этом не рассматриваются возможные аномалии вследствие делеций (см.), расщепления полипептидных цепей и др.
Возникновение Гемоглобинопатий обусловлено мутацией на уровне структурных или регуляторных генов, управляющих синтезом полипептидных цепей глобина. Мутации на уровне структурных генов характерны для истинных Г.: серповидноклеточная анемия, С-, D-, E- и М-гемоглобинопатии и заболевания, обусловленные наличием нестабильных гемоглобинов. Мутации на уровне регуляторных генов обнаруживаются при талассемиях, гемоглобинопатиях H и Bart.
У гомозиготных носителей генов обычно продолжительность жизни эритроцитов с патологическим гемоглобином укорочена, а в некоторых случаях нарушен и эритропоэз. Гетерозиготные носители аномальных гемоглобинов, кроме гемоглобинов H и Bart, вариантов M-гемоглобинопатий, а также носители нестабильных гемоглобинов в обычных условиях жизни являются практически здоровыми людьми.
Большинство Г. наследуется в соответствии с законами Менделя по кодоминантному аутосомному типу, не сцепленному с полом.
Классификация гемоглобинопатий
Классификация гемоглобинопатий [по Гюнсу (E. R. Huehns) в модификации Ю. Н. Токарева] создана на основе результатов специальных биохимических и генетических исследований.
I. Гемоглобинопатии, обусловленные аномалией первичной структуры молекулы гемоглобина («качественные» Г.): 1) серповидноклеточная болезнь, ее варианты (S-гемоглобинопатии: S-талассемия, SD, SC и др.); 2) Lepore-гемоглобинопатия, возникающая вследствие расщепления частей бета- и дельта-цепей глобина; 3) Г. гомозиготные (С-, D- и Е-); 4) M-гемоглобинопатии; 5) Г., обусловленные наличием нестабильных гемоглобинов (гемоглобинов, неустойчивых к воздействию окислителей, нагреванию и др.); 6) Г. бессимптомные (G-гемоглобинопатии и др.).
II. Гемоглобинопатии, вызванные нарушением синтеза полипептидных цепей гемоглобинов («количественные» Г., или талассемии): 1) Г., вызванные нарушением синтеза α-цепи глобина (альфа-талассемия и заболевания, обусловленные наличием гемоглобинов H и Bart); 2) Г., вызванные нарушением синтеза β и δ-цепей глобина (β-талассемия, β,δ-талассемия); 3) бессимптомное наследственное персистирование фетального гемоглобина, т. е. генетически обусловленное увеличенное содержание гемоглобина F у взрослых.
III. Смешанная группа — двойные гетерозиготные состояния по гену талассемии и гену одной из «качественных» Г.
Клиническая картина
Клиническая картина отличается выраженным полиморфизмом. Для серповидноклеточной анемии (см.) и талассемии (см.) характерно развитие тяжелой гемолитической анемии. Ряд известных сейчас Г. клинически не проявляется; варианты гемоглобина при них скорее являются необычными (семейными), чем патологическими.
Lepore-гемоглобинопатия клинически сходна с талассемией.
Гомозиготная гемоглобинопатия C (CC) характеризуется легкой гемолитической анемией со спленомегалией, умеренной желтухой и билирубинемией, нормобластной гиперплазией костного мозга. В периферической крови множество мишеневидных клеток и выраженные морфологические изменения эритроцитов (анизо- и пойкилоцитоз, полихроматофилия), аномальный гемоглобин С составляет св. 90%, нормальный гемоглобин А отсутствует, содержание гемоглобина F в пределах нормы или несколько увеличено (5—7%). Гемолитические кризы с анемией возникают чаще у беременных женщин.
Гомозиготная гемоглобинопатия D (DD) протекает бессимптомно. В единичных случаях наличие аномального гемоглобина D (DD) может проявляться легкой анемией, микросфероцитозом эритроцитов, повышением осмотической резистентности, укорочением продолжительности жизни эритроцитов.
Гомозиготная гемоглобинопатия E (ЕЕ) по клиническому течению очень сходна с гомозиготной гемоглобинопатией С.
У больных М-гемоглобинопатиями ведущим клиническим признаком является врожденный цианоз вследствие повышенного метгемоглобинообразования (см. Метгемоглобинемия).
Для большинства вариантов Г., обусловленных наличием нестабильных гемоглобинов, характерны хроническая несфероцитарная гемолитическая анемия с периодическим выделением темной мочи (вследствие наличия в ней дипирролов) и выраженный полиморфизм клин, проявлений — от бессимптомных до тяжелых форм.
В мазках периферической крови — гипохромия и морфол, изменения эритроцитов — анизо- и пойкилоцитоз, мишеневидность, наличие телец Гейнца и базофильная пунктация (см. Эритроциты). Гемолиз внутриклеточного типа происходит преимущественно в селезенке, что клинически выражается спленомегалией. Другие признаки (ретикулоцитоз, повышение непрямого билирубина и др.) неспецифичны. Г., обусловленные нестабильными гемоглобинами, следует дифференцировать с гемолитическими и гипохромными анемиями другой этиологии и прежде всего с железо дефицитными анемиями. Ценными в диагностике последних являются пробы на термолабильность гемоглобина [Дейси (J. V. Dacie) с соавт., 1964], обнаружение телец Гейнца, выявление в моче дипирролов. Интенсивность аутогемолиза повышена, она уменьшается при добавлении к крови in vitro глюкозы [I тип гемолиза по Селвину (J. G. Selwyn) и Дейси]. Электрофорез гемоглобина не всегда позволяет выявить аномалию, т. к. некоторые нестабильные гемоглобины имеют нормальную (как у гемоглобина А) электрофоретическую подвижность.
Диагноз
Диагноз ставится на основании клинических и лабораторных признаков повышенного гемолиза (см.). Имеет значение выявление морфол, изменений эритроцитов. Подтверждается диагноз данными электрофореза гемоглобина (при структурных Г.: S, С, D, E и др.), пробой на щелочную устойчивость эритроцитов (при подозрении на наличие гемоглобина F), на стабильность гемоглобинов (при подозрении на наличие нестабильных гемоглобинов)
Лечение
Лечение больных Г. в основном симптоматическое, причем выбор метода зависит от степени активности и стадии развития (криз, ремиссия) заболевания. Могут быть применены методы терапии наследственных гемолитических анемий, однако с поправками и дополнениями, обусловленными особенностями данной разновидности аномального гемоглобина (изменения в обмене железа, наличие у некоторых больных гемолитического синдрома с признаками гиперспленизма). При Г. в период криза необходим постельный режим, диета, богатая белками и витаминами; гемотрансфузии (предпочтительна эритроцитарная масса) назначают при концентрации гемоглобина ниже 8—9 г%. Введение в организм препаратов, связывающих и экскретирующих железо (десферал и др.), целесообразно для профилактики гемосидероза.
Вопросы лечения гемолитических анемий, обусловленных нестабильными гемоглобинами, недостаточно изучены. Эффективны иногда стероидные гормоны. Показаниями к спленэктомии являются рефрактерность к обычным методам терапии, тяжелое течение заболевания с выраженным гемолизом, признаки гиперспленизма и секвестрации эритроцитов преимущественно в селезенке. В период ремиссии больные Гемоглобинопатией подлежат диспансерному наблюдению с санацией инфекционных очагов и своевременному лечению интеркуррентных инфекций. Наблюдение за больными должно быть усилено при беременности, в стрессовых ситуациях (хирургическая операция и др.).
Прогноз зависит от вида Г. и клинического течения заболевания.
Профилактика
Необходимы медико-генетические консультации с целью предупреждения браков между гетерозиготными носителями генов аномальных гемоглобинов или с целью предупреждения (при согласии родителей) рождения больного ребенка. Согласно рекомендациям ВОЗ в районах, эндемичных по аномальным гемоглобинам, должны быть созданы специализированные клиники (диспансеры) с медико-генетическими консультациями. Такие центры в СССР созданы при институтах гематологии и переливания крови (Москва, Баку, Ташкент, Тбилиси).
Гемоглобинопатии у детей протекают тяжело, особенно гомозиготные формы — серповидноклеточная анемия, большая талассемия. Диагностируются начиная с шестимесячного возраста, т. е. в период замены гемоглобина F гемоглобином А.
Физическое развитие больных Г. детей замедляется. Характерен их внешний вид, обусловленный изменением костей и увеличением живота за счет гепатоспленомегалии.
Лечение
Дети, больные серповидноклеточной анемией, нуждаются в гемотрансфузиях при уменьшении уровня гемоглобина ниже 8— 9 г%. Больным талассемией переливают эритроцитарную массу даже при незначительном уменьшении содержания гемоглобина.
Больные Гемоглобинопатией дети подлежат диспансерному наблюдению гематолога, плановой иммунизации, им профилактически вводят гамма-глобулин й назначают препараты фолиевой к-ты для предупреждения ее дефицита в организме. В лечении (и профилактике кризов) Г. у детей большое значение имеет осведомленность родителей о природе Г. и их контакт с врачом.
Прогноз
Без надлежащего ухода и лечения большинство больных детей погибает в возрасте 2—5 лет. Особенно тяжело протекают Г. при сочетании их с нарушениями питания (белково-калорийная недостаточность, Квашиоркор и др.) и присоединении интеркуррентных инфекций (корь, пневмония, острые респираторные заболевания).
Библиография: Алексеев Г. А. и Токарев Ю. Н. Гемоглобинопатии, М., 1969; Эфроимсон В. П. Иммуногенетика, с. 161, М., 1971; Beale D. a. Lehmann H. Abnormal haemoglobins and the genetic code, Nature (Lond.), v. 207, p. 259, 1965; Hutchison H. E. An introduction to the haemoglobinopathies and the methods used for their recognition, L., 1967; Jonxis J. H. P. a. Huisman Т. H. J. A laboratory manual on abnormal haemoglobins, Oxford, 1968; Lehmann H. a. Huntsman R. G. Man’s haemoglobins, Philadelphia, 1974; Necheles T. F., Allen D. M. a. Finkel H. E. Clinical disorders of hemoglobin, structure and synthesis, N. Y., 1969; Treatment of haemoglobinopathies and allied disorders, techn. rep. ser. № 509, Geneva, WHO, 1972.
Ю. H. Токарев.
Источник