Виды гемоглобина при талассемии
Содержание статьи
абораторная диагностика бета-талассемии — анализы
Лабораторная диагностика бета-талассемии — анализы
При тяжелой талассемии анемия неизменно тяжелая и резко гипохромная, при этом значения гемоглобина колеблятся от 5 до 7 г/100 мл крови, а показатели гематокрита и численности красных кровяных клеток соответственно уменьшаются. Постоянные эритроцита НЕМ и СНЕМ меньше нормы, соответственно недостаточности гемоглобина в эритроцитах. Вместе с тем, показатель РОЭ колеблется в соответствии с недостаточностью фолиевой кислоты и связанным с ней вторичным макроцитозом.
Мазок периферической крови характерен и с самого же начала исследования подсказывает диагноз. Гипохромия резко выражена, ей сопутствует анизопойкилоцитоз высокой степени. Многочисленные точечные и частые капельные гематии и овалоциты, наряду с большим числом красных кровяных телец искаженного очертания и шизоцитов различной формы и величины, в целом характеризуют мазок при анемии Кули.
Звездочкообразные гематии с подвернутыми краями напоминают «мексиканскую шляпу», при этом иногда они макромегалоцитные (12—15 u). В случаях очень низкого показателя гемоглобина отдельные гематии как бы лишены содержания, в связи с чем принимают аспект «эритроцитной тени». Часто отмечается наличие красных кровяных телец с кольцами Cabot, телец Jolly и базофильными точками. Неизменно присутствие эритробластов (5—20/100 лейкоцитов), при этом, после удаления селезенки, их численность в несколько раз больше лейкоцитов (300—500/100 лейкоцитов).
Выраженная полихроматофилия мазка отражает повышенный ретикулоцитоз (5—15%).
При легкой талассемии анемия умеренная (гемоглобин от 7 до 10 г/100 мл) или небольшая, причем в формах отягчения могут наблюдаться нормальные значения гемоглобина. Степень эритроцитной беспорядочности значительно меньше, чем при тяжелой талассемии. Мазок периферической крови отражает общий аспект микроцитной гипохромной анемии. Тем не менее пойкилоцитоз, наличие звездочкообразных и точечнобазофильных красных кровяных телец делает возможным отличить это заболевание от однородной микроцитно-гипохромной картины железодефицитной анемии.
Небольшая полихроматофилия соответствует слегка увеличенной численности ретикулоцитов (2—5%). Эритробласты редкие (1—2 на несколько сот лейкоцитов) или, как это бывает в большинстве случаев, просто отсутствуют.
Умеренный лейкоцитоз постоянное явление при тяжелой талассемии (10 000—20 000/мм3), ему нередко сопутствует отклонение влево лейкоцитной формулы. У большинства больных наблюдается и небольшой рост показателя численности тромбоцитов. В принципе, при легких формах, число лейкоцитов и тромбоцитов укладывается в норму или у высшего предела.
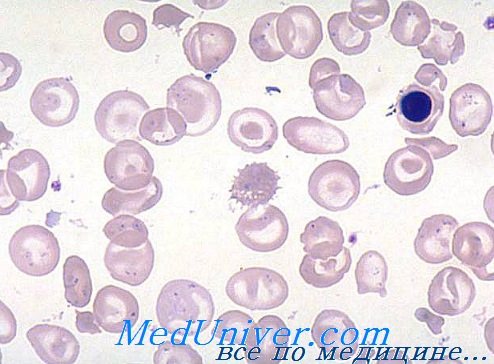
Мазок при бета-талассемии
Косвенный билирубин увеличен (1—3 мг/100 мл), показатели уробилиногена в моче и выделения стеркобилиногена с испражнениями — весьма высокие. При легкой талассемии все эти признаки разрушения красных кровяных клеток существуют, но в значительно сокращенном виде в зависимости от клинической формы и степени расплавления крови.
Осмотическая устойчивость эритроцитов в гипотонном солевом растворе почти неизменно повышена, общее расплавление крови часто отмечается при концентрации примерно 0,2 г NaCl/100 мл или даже меньше в случае выраженного платицитоза. Для диагностирования талассемии реакция весьма полезна, но отнюдь не патогномонична. Отмечаются случаи железодефицитиой анемии с высокой осмотической устойчивостью, подобно случаям талассемии-отягчение, при которой показатели близки к норме.
Сидервмия, общая способность связывания сидерофилина (СС), латентная способность (ЛС), коэффициент насыщения (КН) сидерофилина железом выявляют степень нагрузки организма железом. В большинстве случаев тяжелой талассемии СС меньше нормы (300—330 мкг), показатель сидеремии всегда повышен и КН колеблется от 60 до 100%, подобно тому как наблюдается при гемохроматозе. Окраска на определение костномозгового гемосидерина выявляет увеличенное количество последнего в макрофагах и значительный процент сидеробластов.
При легкой талассемии сидеремия несколько повышена или нормальна, причем в отдельных случаях показатель СС несколько занижен а значения КН сидерофилина укладываются в перделы от 33 до 60%. Вообще процент сидеробластов превышает норму. Определение сидеремии, СС, КН и костномозгового железа особо полезны при дифференциальной диагностике с гипохромной железодефицитиой анемией.
В условиях тяжелой талассемии костный мозг отличается выраженной гиперплазией с явным преобладанием эритробластического ряда (Г/Э = 1/4—1/8). В случае недостатка фолиевой кислоты некоторые эритробласты приобретают мегалобластовидный аспект. При легкой талассемии костномозговой мазок выявляет наличие обильной клеточной массы с умеренным преобладанием эритробластического ряда. В принципе исследование костного мозта не обязательно для постановки диагноза, тем не менее оно полезно для определения запасов железа, в основном при легкой талассемии с электрофорезом гемоглобина в пределях нормы или при осложнениях с хронической кровоточивостью и недостатком железа.
Электрофорез гемоглобина основной способ для уточнения диагноза бета-талассемии. Тяжелая талассемия или анемия Кули характеризуется весьма высоким показателем гемоглобина F (N > 2% у детей старше 1 года), составляющим 20—90% всего гемоглобина, в остальной части отмечается наличие гемоглобин А и нормальные или завышенные значения гемоглобин А2. Наиболее часто значения гемоглобин F составляют 30—60%. В случаях гомозиготной бета-талассемии вида дельта-бета все количество гемоглобина состоит лишь из вида F, при полном отсутствии видов А и А2. Значения Гб в результате электрофореза следует сопоставить со значениями химического теста на определение щелочноустойчивого гемоглобина, в целях исключения аномального гемоглобина, электрофоретическая миграция которого может оказаться одинаковой с гемоглобином F.
Гетерозиготная бета-талассемия характеризуется ростом показателя гемоглобина А2 и/или гемоглобина F, в процентном выражении. Большинство случаев гетерозиготной талассемии (примерно 90%) относятся к виду I при этом показатель гемоглобина А2 колеблется в пределах от 4 до 6% (N > 3%). Значения от 3 до 4%, в частности от 3 до 3,5% следует толковать осторожно и сопоставлять с результатами остальных исследований. Примерно в 50% случаев гетерозиготной бета-талассемии, с высоким показателем гемоглобина А2, отмечается и слегка повышенный показатель гемоглобина F (2—5%), причем для их определения применяется тест на устойчивость к щелочам.
Бывают случаи легкой бета-талассемии, осложняющиеся недостатком железа (повторные роды, хроническая кровоточивость и пр.), при которых значения гемоглобина А2 при электрофоретическом исследовании укладываются в норму. После проведения железотерапии повторный электрофорез выявляет рост показателя гемоглобина А2 за пределы нормы.
Случаи гетерозиготной бета-талассемии, характеризующиеся ростом лишь гемоглобина F редки (среди нашего населения примерно 10% всех гетерозиготов), при этом соответствующий показатель составляет примерно 2—30%, наиболее часто 5—15%; у подобных больных гемоглобин А2 нормальный или заниженный, остальную долю — составляет гемоглобин А. Такие случаи следует отличать от физиологических или патологических состояний с высоким показателем гемоглобин F, таких как, беременность, бирмеровская и сидероакрестическая анемия, лейкемия и пр.
Диагностирование бета-талассемии относительно несложное. Проявление болезни в детстве, клинические и гематологические признаки тяжелой гемолитической анемии подсказывают диагноз наследственной гемолитической анемии. Аспект мазка периферической крови исключает остальные виды наследственной гемолитической анемии, в первую очередь врожденного сфероцитоза. Электрофорез и определение щелочноустойчивого гемоглобина выявляют весьма высокий показатель гемоглобина F, и тем самым подтверждают диагноз.
При легкой форме бета-талассемии клиническое и гематологическое исследования менее убедительны. Наиболее частая ошибка заключается в определении заболевания как гипохромная железодефицитная анемия. Однако пойкилоцитоз, звездочкообразные красные кровяные тельца, базофильноточечные гематии, равно как и признаки расплавления крови свидетельствуют о наличии легкой формы талассемии.
Высокие показатели сидеремии и костномозгового гемосидерина лишний раз подтверждают это предположение. Диагноз подтверждается электрофорезом и определением щелочноустойчивого гемоглобина, поскольку эти исследования выявляют высокие показатели гемоглобина А2 и/или гемоглобина F. Исследования в семейном плане подтверждают наследственный характер заболевания. Так, при тяжелой талассемии, как у отца, так и у матери выявляется наличие легкой талассемии, в то время как при легкой форме этого заболевания, по меньшей мере у одного из них.
— Также рекомендуем «Механизмы развития бета-талассемии — патофизиология»
Оглавление темы «Талассемии»:
- Талассемические синдромы — история изучения, классификация
- бета-Талассемия (анемия Кули) — причины, классификация
- Клиника бета-талассемии — признаки
- Лабораторная диагностика бета-талассемии — анализы
- Механизмы развития бета-талассемии — патофизиология
- Течение бета-талассемии (анемии Кули) — осложнения
- Лечение бета-талассемии — анемии Кули
- а-Талассемия — причины, диагностика, классификация
- Легкие талассемические и талассемическоподобные синдромы: дельта- и гамма-талассемии, синдром Lepore
- Анемии вследствие энзимопатий: недостаток глюкозо-6-фосфатдегидрогеназы (Г-6-ФД) в эритроцитах
Источник
Талассемия – виды, описание. Альфа-талассемия, бета-талассемия, их типы, симптомы талассемии. Гемоглобинопатия. Наследственное персистирование фетального гемоглобина (НПФГ)
Понятие «талассемия» объединяет ряд генетических заболеваний крови. Чтобы понять, что представляет собой талассемия, необходимо понять, как устроена кровь. Гемоглобин является компонентом, переносящим кислород в эритроциты. Он состоит из двух различных белков — альфа и бета. Если организм человека не производит достаточное количество какого-либо из этих белков, красные кровяные клетки не формируются должным образом и не могут переносить достаточное количество кислорода. В результате у человека развивается анемия, которая может начаться в раннем возрасте и продолжается в течение всей жизни.
- Альфа-талассемия, типы альфа-талассемии
- Бета-талассемия, типы бета-талассемии
- Другие формы талассемии
- Симптомы талассемии
- Гемоглобинопатия
- Наследственное персистирование фетального гемоглобина (НПФГ)
Талассемия — это группа родственных расстройств, которые имеют ряд общих и ряд отличительных признаков. Различия между типами талассемии обусловливают те симптомы, которые испытывает человек при развитии этого заболевания.
Из-за преимущественного распространения заболевания среди средиземноморского контингента, талассемию, а точнее различные её виды, называют средиземноморской анемией.
Альфа-талассемия
Люди, чей гемоглобин не производит в достаточном количестве альфа-белок, страдают от альфа-талассемии.
Существует несколько типов альфа-талассемии:
1. Бессимптомный.
Это состояние, как правило, не вызывает никаких проблем со здоровьем, поскольку уровень отсутствия альфа-белка очень мал. Функция гемоглобина при этом не нарушается. Человек, больной таким заболеванием, называется «молчаливым носителем». У таких людей могут родиться дети с признаками альфа-талассемии.
2. Мутация альфа-гемоглобина.
Наблюдается, в основном, на территории Ямайки, где впервые было обнаружено это состояние крови. Проблем со здоровьем у таких пациентов обычно нет.
3. Мягкая форма альфа-талассемии.
В этом состоянии уровень отсутствия альфа-белка несколько выше. Пациенты с такими симптомами имеют умеренную форму альфа-талассемии, ошибочно принимаемую многими врачами за железодефицитную анемию.
4. Синдром Н-гемоглобина.
В этом состоянии у пациентов наблюдаются серьезные проблемы со здоровьем, например, увеличение селезенки, вирусные инфекции. Эта форма заболевания названа в честь аномального гемоглобина Н, который разрушает красные кровяные клетки.
5. Хронический Н-гемоглобин.
Более опасный для здоровья человека синдром, приводящий к появлению серьезной анемии, проблемам с внутренними органами.
6. Гомозиготный Н-гемоглобин.
Этот симптом наблюдается у детей, чьи родители являются носителями хронической формы Н-гемоглобина.
Еще одна форма альфа-талассемии называется водянкой плода. При этом состоянии у новорожденного отсутствуют в ДНК альфа-гены, которые трансформируют гамма-глобины, произведенные организмом, в гемоглобин Барта.
Большинство детей с таким состоянием умирают до или сразу после рождения. В некоторых, крайне редких случаях, внутриутробное переливание крови позволяют ребенку с водянкой родиться живым, однако таким детям требуется пожизненное переливание крови и медицинская помощь.
Бета-талассемия
Люди, чей гемоглобин не производит достаточное количество бета-белка, болеют бета-талассемией. Заболевание чаще встречается у людей средиземноморского происхождения, например, итальянцев, греков, а также обитателей Аравийского полуострова, Ирана, Африки, Юго-Восточной Азии и южной части Китая.
Существует три типа бета-талассемии, которые варьируются от легкой до тяжелой степени, в зависимости от их воздействия на организм.
Типы бета-талассемии
1. Малая бета-талассемия.
Отсутствие бета-белка недостаточно велико, чтобы вызвать проблемы с нормальной функцией гемоглобина. Человек с этим состоянием просто является генетическим носителем признаков талассемии, и с точки зрения врачей практически здоров. Возможна умеренная степень железодефицитной анемии.
Как и при мягкой форме альфа-талассемии, врачи часто ошибочно принимают небольшие эритроциты с бета-талассемией за проявление железодефицитной анемии и назначают препараты железа.
2. Промежуточная талассемия.
Достаточно большой уровень отсутствия бета-белка в гемоглобине, что вызывает анемию умеренной степени, деформацию костей, увеличение селезенки.
3. Бета-талассемия третьей степени (анемия Кули).
Самая тяжелая форма бета-талассемии, при которой в гемоглобине полностью отсутствует бета-белок. Такая форма заболевания является условием для регулярных переливаний крови. Такие обширные переливания крови приводят к перенасыщению железом. Это является условием для хелатотерапии, целью которой является предотвращение ранней смерти от полиорганной недостаточности.
Другие формы талассемии
1. Дельта-бета-талассемия — еще одна форма бета-талассемии, характеризующаяся полным отсутствием или понижением уровня синтеза дельта и бета-цепей глобина с компенсаторным увеличением экспрессии синтеза гамма-цепей. Распространенность этой формы заболевания неизвестна, однако чаще всего она встречается в Греции и Италии.
Гетерозиготная форма заболевания протекает клинически бессимптомно, с мягким микроцитозом и повышением уровня гемоглобина HbA2. При гетерозиготной наследственной бета-талассемии у больных производится только гамма-глобин с образованием HbF.
2. E-бета-талассемия.
Гемоглобин Е является одним из наиболее распространенных аномальных гемоглобинов. Заболевание проявляется у жителей Юго-Восточной Азии, например, камбоджийцев, вьетнамцев и тайцев. В сочетании с бета-талассемией, гемоглобин Е вызывает умеренно-тяжелую анемию, которая схожа с симптомами промежуточной бета-талассемии.
3. Серповидная-бета-талассемия.
Обусловлена сочетанием бета-талассемии и гемоглобина S, аномального гемоглобина, свойственного людям, больным серповидно-клеточной анемией. Это заболевание характерно для греков, турков, итальянцев. Чем больше бета-глобина производит бета-ген, тем тяжелее заболевание и его симптомы.
Симптомы талассемии
Талассемия в слабой степени обычно не вызывает никаких симптомов. Если всё же симптомы и есть, они напоминают мягкую форму анемии.
Наиболее выражены симптомы анемии Кули. Они проявляются на второй год жизни ребенка. Кроме проявлений железодефицитной анемии, возможны такие симптомы:
- частые вирусные инфекции;
- бледность;
- плохой аппетит;
- желтуха;
- увеличение внутренних органов.
Гемоглобинопатия
Гемоглобин производится генами, которые контролируют экспрессию этого белка. Дефекты в этих генах могут производить аномальные типы гемоглобина и приводят к анемии, которую называют гемоглобинопатия. Аномальные типы белка-гемоглобина появляются в таких случаях:
1. Структурные дефекты в молекуле гемоглобина.
Случается так, что мутации изменяют одну из аминокислот. Чаще всего такое изменение безобидно и не нарушает ни структуру, ни функцию молекул гемоглобина. Однако случается и так, что изменение одной аминокислоты резко нарушает поведение молекулы гемоглобина и приводит к болезням.
2. Уменьшение производства одной из двух субъединиц молекул гемоглобина.
Равные количества гемоглобина альфа- и бета-цепей являются необходимыми для нормальной функции крови. Дисбаланс в цепи гемоглобина повреждает и разрушает эритроциты, тем самым приводя к анемии.
3. Аномальные соединения нормальных субъединиц.
Одна субъединица альфа-цепи и одна субъединица бета-цепи объединяются, чтобы произвести нормальный гемоглобин. В тяжелых случаях субъединицы глобина начинают объединяться в группы по четверо. Они неактивны функционально и не транспортируют кислород. При этом альфа-субъединицы быстро деградируют при отсутствии партнера типа бета.
Наследственное персистирование фетального гемоглобина (НПФГ)
Персистенция (персистирование) фетального гемоглобина (HPFH, НПФГ) является состоянием, при котором производство фетального гемоглобина (гемоглобина F), продолжается и в зрелом возрасте.
Фетальный гемоглобин — это основной вид гемоглобина, который вырабатывается у плода во время нахождения в утробе матери. Наличие такого элемента в организме позволяет при достаточно малом количестве крови в организме плода выполнять кислородоснабжающую функцию.
Когда ребенок рождается, гемоглобин-Ф заменяется гемоглобином-А, его более «взрослой» формой. Если же замена происходит не полностью, это свидетельствует от талассемии.
Причины
НПФГ вызвана мутациями в генах β-глобина. Процент таких мутаций обычно равен 10-15%. В основном это состояние бессимптомно и обнаруживается во время скрининга, причиной которого является другое расстройство крови.
Источники статьи:
https://www.nhlbi.nih.gov
https://www.thalassemia.org
https://www.healthline.com
https://www.orpha.net
https://en.wikipedia.org
По материалам:
1.Pr Renzo GALANELLO, Dr Raffaella ORIGA
2.Gretchen Holm, George Krucik, MD
3.The Cooley’s Anemia Foundation
4.National Institutes of Health, Department of Health and Human Services
5.wikipedia.org
Смотрите также:
У нас также читают:
Источник
Гемоглобинопатия — аномальные варианты гемоглобина | Университетская клиника
Гемоглобинопатия – это наследственные заболевания с единой проблемой – образованием аномальной формы гемоглобина, например, серповидноклеточная анемия S и талассемия.
Гемоглобинопатии носят эндемический характер – они возникают в определенном географическом районе, например, в Средиземноморье, Африке, Юго–Восточной Азии. В нашей стране они тоже встречаются.
Что такое гемоглобинопатия
Гемоглобинопатии – это заболевания, вызванные выработкой и присутствием аномальной формы гемоглобина.
Гемоглобин состоит из гема (частей, содержащих железо) и глобина (частей белка, состоящих из аминокислотных цепей). Молекулы гемоглобина (Hb или Hgb) находятся в красных кровяных тельцах. Их задача – связывать кислород в легких и передавать его тканям и органам, где они его выделяют.
Строение гемоглобина
Существует несколько типов цепей глобина: альфа, бета, дельта и гамма.
Типы нормального гемоглобина:
- A – HbA: составляет около 95-98% от общего гемоглобина у взрослых людей. Он содержит 2 альфа (α) цепи и две бета (β) цепи.
- A2 – HbA2: составляет около 2-3% от общего гемоглобина. Он содержит 2 цепи альфа (α) и две цепи дельта (δ).
- F (HbF): составляет около 2% от общего гемоглобина взрослого человека. Он содержит 2 альфа (α) цепи и две гамма (γ) цепи. Этот гемоглобин в основном вырабатывается у плода, его производство значительно снижается вскоре после рождения и достигает уровня взрослого человека в течение 1-2 лет.
К гемоглобинопатиям относятся: структурные варианты гемоглобина, гемоглобин S, серповидноклеточная анемия, гемоглобинопатия C, гемоглобинопатия E, талассемия, гемоглобин Бартс, наследственная персистенция гемоглобина плода.
Причины развития гемоглобинопатии
Гемоглобинопатии возникают в случае генетических изменений генов глобина, которые приводят к изменению аминокислот, составляющих белок глобина. Эти изменения влияют на:
- структуру гемоглобина, например, гемоглобин S, который вызывает серповидно-клеточную анемию;
- его поведение;
- количество продуцируемого вещества (талассемия);
- стабильность.
Серповидно-клеточная анемия
Существует четыре гена, кодирующих цепь альфа-глобина, и два гена, кодирующих цепь бета-глобина. Наиболее частым заболеванием, связанным с изменением альфа-цепи, является альфа-талассемия. Его тяжесть зависит от количества пораженных генов.
Талассемия характеризуется снижением продукции одной из цепей глобина, дисбалансом между альфа- и бета-цепями в гемоглобине A (альфа-талассемия) или увеличением малых форм, таких как Hb A2 или Hb F (бета-талассемия).
Изменения бета-цепей гемоглобина являются врожденными, аутосомно-рецессивными. Это означает, что больной человек должен иметь две дефектные копии гена, каждая от одного из родителей. Если один ген нормален, а другой дефектен, человек гетерозиготен, и мы называем его носителем. Аномальный ген может быть передан любому из потомков. Если рассматриваемый человек является гетерозиготным носителем, он может не иметь никаких симптомов и носительство не влияет на его здоровье.
Если происходят две модификации одного и того же бета-гена, человек гомозиготен по этому гену. Его организм может производить дефектный гемоглобин – возникает гемоглобинопатия с симптомами и потенциальными осложнениями. Степень тяжести зависит от генетической мутации и варьируется от случая к случаю. Копию гена можно передать потомству.
Если два аномальных бета-гена являются врожденными, человек является двойным, смешанным гетерозиготным. У него будут симптомы одной или обеих гемоглобинопатий. Один из аномальных бета-генов будет передаваться каждому из потомков.
Были идентифицированы сотни гемоглобинопатий в бета-цепях. Хотя лишь некоторые из них являются общими и клинически значимыми.
Клинические признаки и симптомы
Признаки и симптомы различаются по типу гемоглобинопатии и возможному сочетанию нескольких гемоглобинопатий. Некоторые приводят к усилению распада эритроцитов (гемолизу), уменьшению их общего количества и развитию анемии.
Клинические признаки включают:
- слабость, утомляемость;
- недостаток энергии;
- желтуха;
- бледность кожи.
Утомляемость
К серьезным клиническим признакам относятся:
- приступы сильной боли;
- удушье;
- увеличение селезенки;
- нарушения роста у детей;
- боль в верхней части живота (вызванная желчными камнями).
Удушье
Общие гемоглобинопатии
Красные кровяные тельца, содержащие аномальный гемоглобин, могут не переносить кислород достаточно эффективно. Они могут разрушаться раньше (чем в здоровых клетках крови) и развиваться гемолитическая анемия. Выявлены сотни гемоглобинопатий, но лишь некоторые из них являются общими и клинически значимыми.
Одной из наиболее распространенных гемоглобинопатий является серповидно-клеточная анемия с присутствием гемоглобина S. Это приводит к изменению формы – серповидно-клеточной – эритроцитов и снижению их выживаемости. Гемоглобин С может вызвать легкую гемолитическую анемию. Гемоглобин E обычно не приводит к развитию каких-либо или только очень легких клинических симптомов.
- Талассемия: самая распространенная генетическая аномалия в мире. Она часто встречается в Средиземноморье, на Ближнем Востоке и в Юго-Восточной Азии. Более легкая форма талассемии также встречается, например, у людей, родившихся в Чехии.
- Гемоглобин S: это основной гемоглобин людей с серповидно-клеточной анемией. В среднем эта мутация есть в одном из двух бета-генов у 8% американцев и африканцев. Возникновение этих мутаций в наших широтах встречаеся редко. Пациенты с заболеванием HbS имеют две аномальные цепи бета (b s) и две нормальные цепи альфа (a). Когда эритроциты, содержащие гемоглобин S, подвергаются действию пониженного количества кислорода (как это может быть в случае повышенной физической нагрузки или инфекционного заболевания легких), они деформируются, принимая форму полумесяца. Серповидные эритроциты могут блокировать периферические кровеносные сосуды и вызывать нарушения кровотока и боль. У них пониженная способность переносить кислород и более короткий срок жизни. Одна копия б не вызывает клинических проявлений, если не сочетается с другой мутацией гемоглобина, такой как HbC (b C) или бета-талассемия.
- Гемоглобин C: около 25% жителей Западной Африки и 2-3% афроамериканцев гетерозиготны по гемоглобину C (у них есть одна копия B C). Но заболевают только гомозиготные люди с обоими дефектными генами (b C). Обычные симптомы – легкая гемолитическая анемия с небольшим или средним увеличением селезенки.
- Гемоглобин E: вторая по распространенности гемоглобинопатия в мире с изменением бета-цепей. Патология очень часто встречается в Юго-Восточной Азии, особенно в Камбодже, Лаосе и Таиланде, а также частично в Северо-Восточной Азии. Есть случаи и в нашей стране. Люди с гомозиготным Hb E (две копии b E) обычно имеют легкую гемолитическую анемию, микроциты (маленькие красные кровяные тельца) и слегка увеличенную селезенку. Одна копия гемоглобина E не вызывает клинических признаков, если не сочетается с другой мутацией, такой как одна из бета-талассемии.
Талассемия
Необычные гемоглобинопатии
Существует ряд гемоглобинопатий, некоторые из которых не проявляются – они не вызывают никаких клинических признаков или симптомов. Другие, в свою очередь, влияют на функциональность и / или стабильность молекулы гемоглобина. Примерами являются гемоглобин D, гемоглобин G, гемоглобин J, гемоглобин M и гемоглобин Constant Spring. Мутации в гене альфа-цепи глобина приводят к образованию аномально длинных альфа (а) цепей, которые вызывают нестабильность в молекуле гемоглобина.
Другие примеры мутаций бета-цепи:
- Гемоглобин F: Hb F в основном вырабатывается в организме будущего ребенка (плода), и его функция заключается в переносе кислорода в среде с низким содержанием кислорода. Продукция гемоглобина F снижается сразу после рождения и стабилизируется на уровне взрослого человека до 1-2 лет. Гемоглобин F может быть повышен при некоторых врожденных заболеваниях. При бета-талассемии его уровень может быть нормальным или повышенным, но часто повышен при серповидно-клеточной анемии и сочетании серповидно-клеточной анемии с бета-талассемией. Пациенты с серповидно-клеточной анемией и повышенным Hb F часто имеют более легкое течение болезни, поскольку Hb F предотвращает серповидное движение красных кровяных телец. Уровни Hb F повышены в редком состоянии, называемом врожденным постоянством выработки гемоглобина плода (HPFH). Люди с повышенным уровнем гемоглобина F не имеют клинических признаков. HPFH вызывается разными генными мутациями у разных этнических групп. Hb F также может быть повышен при некоторых приобретенных состояниях, влияющих на выработку красных кровяных телец. Например, лейкемия и миелопролиферативные заболевания часто сопровождаются небольшим повышением уровня гемоглобина F.
- Гемоглобин H: HbH – это аномальный гемоглобин, который возникает в некоторых случаях альфа-талассемии. Его образование является ответом на фундаментальный недостаток альфа (а) цепей. Hb H состоит из четырех цепей бета (b) глобина. Хотя каждая из цепей бета-глобина нормальна, комплекс из четырех цепей бета нормально не функционирует. Обладает повышенным сродством к кислороду, плохо выделяет кислород клеткам тканей. Присутствие гемоглобина H также связано со значительным распадом эритроцитов (гемолизом), который возникает в результате осаждения нестабильного гемоглобина внутри красных кровяных телец.
- Hemoglobin Barts: Hb Barts вырабатывается в организме будущего ребенка с альфа-талассемией при условии, что все четыре гена, отвечающие за производство гемоглобина альфа, отсутствуют. Таким образом, не может образовываться гемоглобин HbA, HbA 2 и HbF. Гемоглобин Бартс состоит из четырех гамма (g) цепей и имеет высокое сродство к кислороду. Это состояние несовместимо с жизнью и обычно приводит к внутриутробной гибели плода.
Некоторые люди могут унаследовать два гена с разными мутациями, каждый от одного из родителей. Таких людей называют двойными или смешанными гетерозиготами.
Обследование: лабораторные тесты
Исследование на гемоглобинопатию проводится в следующих случаях:
- Выявление гемоглобинопатий у бессимптомных родителей больных детей.
- Выявление гемоглобинопатий у пациента с необъяснимой анемией, микроцитозом и / или гипохромией. Анализ может быть выполнен как часть теста на анемию.
- Скрининг на гемоглобинопатии у новорожденных – только в США и некоторых регионах с повышенной заболеваемостью.
- Пренатальный скрининг проводится в некоторых регионах с высокой частотой гемоглобинопатий (особенно в Африке).
На результаты тестов на гемоглобинопатию может повлиять переливание крови. Поэтому после переливания крови, прежде чем сдать анализ, пациенту следует подождать несколько месяцев. Тем не менее пациентам с серповидно-клеточной анемией после переливания крови рекомендуется сдать анализ крови, чтобы увидеть, достаточно ли гемоглобина в крови, и снизить риск повреждения организма серповидными эритроцитами.
Обследование гемоглобинопатий основано на обнаружении и оценке «нормальности» эритроцитов и гемоглобина в эритроцитах, а также на исследовании конкретной мутации гена. Каждый из тестов является частью головоломки, предоставляющей важную информацию о том, какая гемоглобинопатия присутствует. Для проверки гемоглобинопатии используются следующие тесты:
- Анализ крови. Анализ крови дает быструю информацию о клетках, циркулирующих в крови. Помимо прочего, результаты анализа крови показывают, сколько красных кровяных телец (эритроцитов) содержится в крови, какого они размера и формы, а также сколько там гемоглобина. Размер эритроцита определяет средний объем эритроцитов (MCV). Обнаружение пониженного MCV (микроцитоз, наличие небольших эритроцитов) часто сначала указывает на возникновение талассемии. Если MCV низкий и дефицит железа исключен, пациенты могут быть носителями талассемии или гемоглобинопатии, которые также вызывают микроцитоз (например, HbE).
- Анализ ДНК. Этот анализ используется для скрининга мутаций и делеций в альфа- и бета-областях глобиновых генов. Иногда обследуются все члены семьи. Задача в том, чтобы определить конкретный тип мутации, встречающейся в семье, и выявить всех носителей. ДНК-тесты не являются обычным тестом, но они могут помочь диагностировать гемоглобинопатию и выявить носителей.
- Мазок периферической крови (микроскопический дифференциальный подсчет лейкоцитов, считываемый по мазку периферической крови). Тест проводится путем формирования тонкого слоя крови на предметном стекле и окрашивания его специальными красителями. Образец крови, обработанный таким образом, затем оценивается лаборантом под микроскопом. Специалист определяет количество и тип белых и красных кровяных телец и тромбоцитов. Оценивает, являются ли они нормальными и зрелыми.
Анализ крови
При гемоглобинопатии эритроциты могут быть в следующих формах:
- Микроциты (меньше нормального).
- Гипохромные (более бледные, с пониженным гемоглобином).
- Разных размеров (анизоцитоз) и формы (пойкилоцитоз, например, серповидно-клеточные клетки).
- С ядром (в незрелых эритроцитах) или с включениями.
- С неравномерным распределением гемоглобина (клетки-мишени, которые под микроскопом выглядят как «бычий глаз»).
Наличие более высокого процента аномально выглядящих эритроцитов означает более высокую вероятность наличия заболевания.
С помощью тестов на гемоглобинопатию и их комбинаций можно диагностировать наиболее распространенные гемоглобинопатии. Эти тесты могут помочь выявить пациентов с сочетанием различных гемоглобинопатий (смешанные гетерозиготы).
Лечение гемоглобинопатии
В настоящее время гемоглобинопатии – неизлечимые заболевания. Но возможно устранять симптомы заболевания. Цель – облегчить боль и минимизировать возможные осложнения. Также существуют лекарства, повышающие уровень гемоглобина F, что облегчает некоторые симптомы.
Однако исследования и поиск более безопасных и эффективных методов лечения все еще продолжается. В будущем для восстановления мутированного гена можно будет использовать трансплантацию стволовых клеток или генную терапию. Для того чтобы эти методы могли широко использоваться в будущем, необходимы дальнейшие обширные исследования.
Источники: БЕРТИС, Калифорния, ЭШВУД, Эр., Брунс, Делавэр, (ред.), Учебник Тиц по клинической химии и молекулярной диагностике. 4-е издание Луи: Эльзевье-Сондерс, 2006; LOTHAR, T. Клиническая лабораторная диагностика. Франкфурт: TH-Books, 1998; MASOPUST, J. Клиническая биохимия – требования и оценка биохимических исследований, часть I. и часть 2, Прага: Каролинум, 1998; RACEK, J., et al. Клиническая биохимия. 2. переработанное издание, Прага: Гален, 2006; Каспер Д.Л., Браунвальд Э., Фаучи А.С., Хаузер С.Л., Лонго Д.Л., редакторы Джеймсон Д.Л., 2005.
Поделиться ссылкой:
Источник