Врожденный холестерин что это
Содержание статьи
Семейная гиперхолестеринемия (Наследственная гиперхолестеринемия, Первичнся гиперхолестеринемия)
Семейная гиперхолестеринемия (СГХС) — наследственная патология, характеризующаяся выраженным повышением количества липопротеинов низкой плотности (ЛПНП) в кровотоке и высоким риском раннего развития ишемической болезни сердца. В большинстве случаев протекает бессимптомно. Иногда отмечается боль в груди, волдыри на руках, коленях и вокруг глаз, сухожильные и подкожные отложения холестерина. Основные методы диагностики — сбор наследственного анамнеза, анализ крови на общий холестерин и ЛПНП. Для лечения используется гиполипидемическая диета, регулярные физические нагрузки, медикаментозная коррекция липидоснижающими препаратами, аферез атерогенных липопротеидов.
Общие сведения
Синонимы семейной гиперхолестеринемии — первичная, наследственная гиперхолестеринемия. Является не самостоятельным заболеванием, а состоянием предрасположенности к сердечно-сосудистым болезням — атеросклерозу сосудов, ИБС, острому инфаркту миокарда. Данные о распространенности СГХС имеют большой разброс, поскольку во многих случаях патология остается недиагностированной. Частота гетерозиготной формы, при которой у пациента имеется один дефектный ген из пары, составляет 1 случай на 108-300 человек. Гомозиготная форма, характеризующаяся наличием двух мутационных генов в аллели, протекает тяжелее и встречается гораздо реже — у 1 человека из 1 миллиона. Среди всех вариантов гиперхолестеринемии на долю семейной ГХС приходится 10% случаев.
Семейная гиперхолестеринемия
Причины
СГХС является наследственной аутосомно-доминантной патологией, которая вызывается мутацией генов, ответственных за метаболизм ЛПНП и активность их рецепторов. При наличии одного дефектного гена в паре возникает гетерозиготная гиперхолестеринемия — легкое и умеренное нарушение метаболизма липидов. В редких случаях у пациентов присутствует два парных измененных гена (от матери и от отца), развивается гомозиготная гиперхолестеринемия — тяжелое расстройство липидного обмена со злокачественным течением. Причиной семейной гиперхолестеринемии является мутация в одном из следующих генов:
- LDLR. Ген задает функциональность ЛПНП-рецептора, расположенного, в основном, на поверхности клеток печени. При мутации его активность снижается, процесс связывания и выведения из кровотока циркулирующих липопротеидов нарушается. Выявлено более 1600 видов мутаций гена LDLR. Их доля в общем количестве СГХС составляет 85-90%.
- APOB. Дефект гена приводит к изменению структуры аполипопротеина B100, входящего в состав ЛПНП, обеспечивающего их связывание с рецептором. Мутационные изменения APOB имеются у 5-10% больных наследственной гиперхолестеринемией. Они провоцируют менее выраженное повышение ЛПНП, чем мутации LDLR.
- PCSK9. Этот ген кодирует фермент пропротеиновую конвертазу субтилизин-кексинового типа 9, которая усиливает разрушение ЛПНП-рецепторов. Мутации в гене PCSK9 увеличивают активность фермента, в результате чего количество рецепторов уменьшается. Патология такого типа встречается в 5% случаев СГХС.
Патогенез
В основе семейной гиперхолестеринемии лежит генетически обусловленное повышение уровня ЛПНП. Чаще всего оно вызывается снижением активности специфического рецептора, ответственного за выведение липопротеинов. ЛПНП — наиболее атерогенные частицы. Атеросклеротические бляшки формируются при их накоплении в субэндотелиальном пространстве. Чем выше уровень липопротеинов с низкой плотностью в крови, тем интенсивнее протекает процесс.
Хуже всего ЛПНП выводятся у людей с гомозиготной первичной гиперхолестеринемией: оба парных гена имеют мутацию, функциональность рецептора снижена на более чем на 50%, концентрация ЛПНП высокая, плохо поддается коррекции при помощи медикаментов и диеты. Атеросклероз и его осложнения развиваются в детском и подростковом возрасте. При гиперхолестеринемии гетерозиготного типа только один ген дефектный, половина или более рецепторов остаются функциональными, количество ЛПНП повышается, но долгое время не проявляется клинически. Зачастую первым признаком СГХС становится атеросклероз, ишемическая болезнь сердца или инфаркт миокарда.
Симптомы
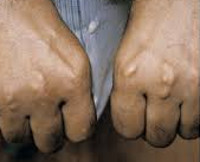
СГСХ развивается с рождения, но часто не имеет выраженных клинических признаков. Диагноз устанавливается с опозданием при манифестации сердечно-сосудистых заболеваний, таких как ИБС, инфаркт сердечной мышцы, атеросклероз. Симптомы гиперхолестеринемии наблюдаются менее чем у половины больных. Примерно у трети пациентов формируются сухожильные ксантомы — уплотнения из жироподобного вещества (холестерина), прощупываемые над сухожилиями. Узелки особенно легко определяются на кистях. Холестерин откладывается под кожей век, возле глаз в виде ксантелазм — желтоватых или не имеющих специфического цвета плоских узелков.
Патогномоничный признак СГСХ — липоидная дуга роговицы. Она представляет собой скопления холестерина по краю роговицы, которые обнаруживаются при офтальмологическом осмотре и выглядят как белый или серо-белый ободок. В отдельных случаях больные отмечают боль и дискомфорт в области груди, водянистые высыпания на коже рук, локтей и коленей. На стадии развития атеросклероза выявляются мозаичные симптомы поражения внутренних органов.
Осложнения
При отсутствии лечения гомозиготная первичная гиперхолестеринемия способствует развитию атеросклероза до 20-летнего возраста, продолжительность жизни больных не превышает 30 лет. Нелеченые пациенты с гетерозиготной формой патологии имеют высокий риск развития ИБС, к 60 годам диагноз подтверждается у 85,5% мужчин и у 53% женщин. Средние показатели продолжительности жизни для мужчин составляют 53 года, для женщин — 62 года. ИБС становится причиной смерти половины мужчин с наследственной гетерозиготной гиперхолестеринемией. Около 20% случаев инфаркта миокарда до 45 лет связаны с наличием СГХС.
Диагностика
Обследованием пациентов занимаются терапевт, кардиолог, врач-генетик. Важным этапом диагностики является сбор личного и семейного анамнеза. Учитывается возраст больного и время начала симптомов, потому что для наследственной патологии характерен ранний дебют. В пользу диагноза семейной гиперхолестеринемии рассматривается наличие двух и более близких родственников (особенно детей) с повышенным холестерином крови, ксантомами и/или липоидными дугами роговиц. Основная задача дифференциальной диагностики — исключение вторичной гиперхолестеринемии. Обследование больных проводится следующими методами:
- Физикальный осмотр. При тщательной пальпации сухожилий стоп, голеней и кистей обнаруживаются ксантомы. На роговице определяется наличие полной или частичной липоидной дуги, у лиц моложе 45-48 лет она указывает на СГХС. Отсутствие ксантом, ксантелазм и роговичной дуги не исключает наличия гиперхолестеринемии.
- Липидограмма. Комплексное лабораторное исследование липидного профиля является наиболее информативным методом диагностики. Показатель общего холестерина при гетерозиготной патологии составляет 7,5-14 ммоль/л, при гомозиготной — 14-26 ммоль/л. Уровень ЛПНП соответственно повышается до 3,3-4,9 ммоль/л и до 4,15-6,5 ммоль/л.
- Генетический скрининг. Выявление мутаций и их характера необходимо при невозможности подтвердить диагноз другими способами, а также для составления оптимального плана лечения. У 80% пациентов обнаруживаются дефекты в генах LDLR, APOB или PCSK9. У оставшихся 20% генетические изменения не диагностируются даже при развернутых симптомах СГХС.
Лечение семейной гиперхолестеринемии
Терапия включает комплекс мероприятий, нацеленных на снижение количества ЛПНП. Тактика определяется формой гиперхолестеринемии, величиной отклонения показателей липидограммы от нормы, выраженностью симптомов и возрастом пациента. Значительная часть лечебных процедур проводится амбулаторно при регулярном контроле эффективности лечащим врачом. Пациентам назначается:
- Медикаментозная терапия. Применяются препараты, понижающие уровень липидов в плазме крови. Наиболее целесообразен комбинированный прием статинов, фибратов, секвестрантов желчных кислот и ингибиторов абсорбции холестерина в кишечнике.
- Коррекция образа жизни. Исключаются все факторы риска гиперлипидемии: требуется полный отказ от курения, контроль артериального давления, нормализация массы тела, регулярная физическая нагрузка. Диетотерапия основана на ограничении количества насыщенных жиров и трансжиров. Ежедневное поступление холестерина с пищей — не более 200 мг.
- Аферез ЛПНП. При гомозиготном типе гиперхолестеринемии медикаментозное лечение зачастую оказывается недостаточно результативным. Проводятся процедуры удаления липопротеинов из крови. Аферез также может быть показан больным ИБС и атеросклерозом с СГХС гетерозиготной формы, особенно если прием лекарств не дает ожидаемого положительного эффекта.
- Стимуляция ЛПНП-рецепторов. С недавних пор в медицинскую практику внедряется патогенетическая терапия СГХС. Используется препарат, стимулирующий увеличение количества рецепторов ЛПНП в клетках печени. В итоге усиливается захват и выведение из организма липопротеинов.
Прогноз и профилактика
Благоприятное течение семейной гиперхолестеринемии наиболее вероятно при гетерозиготном типе, раннем начале лечения и периодическом контроле уровня холестерина на протяжении всей жизни. Из-за наследственного характера патологии предупредить ее развитие невозможно. Профилактические меры нацелены на раннюю диагностику гиперхолестеринемии, что позволяет сократить вероятность атеросклероза, ИБС, инфаркта мышцы сердца. Для этого проводится каскадный скрининг — исследование уровня липидов крови у всех ближайших родственников пациента.
Источник
Высокий холестерин с детства. Семейная гиперхолестеринемия.
ЗАДАТЬ ВОПРОС РЕДАКТОРУ РАЗДЕЛА (ответ в течение нескольких дней)
25 сентября 2009 18:51 | Татьяна Васильева
В медицине существует такое понятие, как семейная гиперхолестеринемия, означающее врожденное наследственное заболевание, характеризующеся повышением уровня холестерина в крови. Для пациентов, страдающих им очень важно вести здоровый образ жизни и находиться под наблюдением у специалиста в терапии внутренних болезней с целью предупреждения тяжелых осложнений со стороны сердечно —
сосудистой
системы.
Это расстройство — я
вление распространенное. На каждые 400 человек найдется случай этого заболевания. Причиной является мутация гена в 19 хромосоме, что блокирует клетки печени, ответственные за удаление холестерина из крови. Больной человек имеет 50% —
ную
вероятность передачи поврежденного гена своим потомкам. В остальных случаях — передается правильная генетическая информация. Поэтому обычно у половины членов одной и той же семьи возникает это расстройство. За того, у кого нормальный уровень холестерина в крови не следует беспокоиться. Он наследовал нормальный ген. Ни у него не разовьется болезнь, ни он ее не передаст потомкам. Исключением из этого правила могут быть 8% пациентов, особенно среди молодых людей, являющихся носителями поврежденного гена, но не имеющих высокого уровня холестерина в крови. Они тоже являются передатчиками болезни последующим поколениям. Поэтому в таких семьях очень важно проведение генетического обследования.
Больные семейной гиперхолестеринемией подвергаются высокому риску развития инфаркта
миокарда в молодом возрасте, а также других атеросклоротичесих поражений сосудов (аневризма аорты, инсульты). Для них исключительно важен ранний диагноз заболевания, лечение и наблюдение врачом- специалистом в терапии. К сожалению большинство таких больных оказывается вне поле зрения медиков и высокий уровень холестерина выявлется у них случайно при профилактическом осмотре или при обращении больного к врачу совсем по другому поводу.
Единственно достоверным методом подтверждения диагноза является результаты генетического обследования. Эти методы основываются на анализе структуры ДНК, поврежденном при этом заболевании гена. Когда диагноз ясен, важно
чтобы пациент понял необходимость лечения и диспансерного наблюдения у врача для предупреждения возможных осложнений.
Терапия этого расстройства включает в себя назначение средств, снижающих уровень холестерина в крови, а также контроль за максимально здоровым образом жизни, который должен поддерживать больной. Этим пациентам необходимо поддерживать нормальный вес, исключить употребления алкоголя и жиров животного происхождения, особенно если наряду с холестерином, увеличен уровень триглицеридов в крови. Им следует заниматься спортом, поддерживать активный образ жизни.
Врач-специалист
в терапии назначает фармакологиеческое лечение всегда с учетом истории болезни каждого больного и наличия сопутствующих заболеваний.
Поделиться:
29 сентября 2009 | 16:09
Лечение гипергидроза. Лечение повышенного потоотделения
Так уж сложилось, что лечением гипергидроза, или, попросту говоря, повышенной потливости, в нашей стране практически никто не занимался. Классическая медицина просто не считала этот вопрос «своим». В самом деле, формально гипергидроз нельзя назвать болезнью, он не снижает работоспособности и не способен привести к тяжелым осложнениям. Но… постоянно и обильно потеющие ноги, ладони, подмышки, лицо могут всерьез отравить жизнь обладателю такой «особенности», а в некоторых случаях стать препятствием к любимой работе, помешать успешной карьере.
23 сентября 2009 | 12:09
Что такое болезнь легионеров, или легионелез?
Возбудитель этого заболевания Legionella Pneumophila, получила свое название из за того, что впервые была обнаружена в конце 70 годов у ветеранов Американского Легиона, собравшихся на очередной съезд . Внезапно у 221 из них развилась тяжелейшая пневмония и 34 человека умерло в больнице.Пневмония, вызываемая легионеллой ,очень тяжелая , с трудом поддающаюся терапии.
21 сентября 2009 | 03:09
Почему «свиной» грипп заражает молодежь?
Так уж получилось, что к новому гриппу, который в медицине получил название гриппа А /N1h2/ прилепилось у нас в народе название «свиного», каким изначально его называли медики. Во-первых, так проще, официальное название — просто не запомнить-не выговорить, во-вторых, чтоб отличить его от «птичьего», который тоже был гриппом А, а в-третьих по большому счету, как грипп не назови, только бы лекарства и вакцины были для защиты, а вот с этим-то как раз — с обеспечением адекватной терапией всех заболевших,вероятее всего во многих странах придется туго.
17 сентября 2009 | 11:09
Гепатит В — скрытый незнакомец
Медицине известно несколько видов вирусных заболеваний печени, среди них гепатит (воспаление печени), вызванный вирусом гепатита В. Его легко обнаружить анализом крови, он излечивается, существует вакцина, предохраняющая от заражения им, и тем не менее число заболеваемости этим гепатитом неуклонно растет. Отсутствие информации о существовании болезни, способе передачи инфекции (чаще всего при сексуальных контактах) и ее терапии во многом объясняет такое положение дел.
15 сентября 2009 | 10:09
Гидротерапия — это лечение водой
Гиппократ, знаменитый греческий лекарь и мыслитель, основатель медицины впервые описал лечебные свойства холодной и горячей воды, ванн, душей, грязевых обертываний, горячих компрессов, которыми и до сих пор пользуются специалисты в курортологии.
Источник
енетика семейной гиперхолестеринемии. Наследование, молекулярные основы
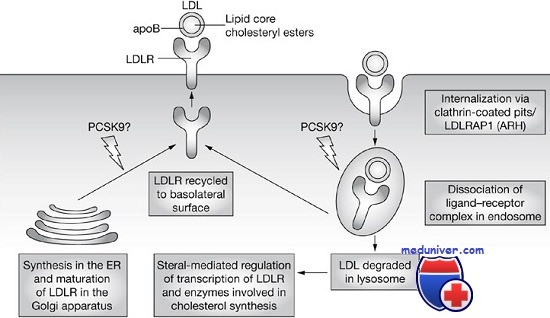
Генетика семейной гиперхолестеринемии. Наследование, молекулярные основыОткрытие класса болезней, вызванных дефектами в молекулах рецепторов, произошло после идентификации в 1974 г. Голдстейном и Брауном рецептора ЛПНП как полипептида, повреждаемого при наиболее частой форме семейной гиперхоле-стеринемии. Это заболевание, приводящее к значительному повышению риска инфаркта миокарда, характеризуется повышением холестерина плазмы, переносимого ЛПНП, главным транспортным белком холестерина в плазме. Открытие Голдстейна и Брауна в общих чертах прояснило нормальный метаболизм холестерина и биологию поверхностных рецепторов клетки. Недостаточность рецептора ЛПНП — первый пример из множества выявленных впоследствии нарушений, вызванных дефектами рецепторов. Семейная гиперхолестеринемия: генетическая гиперлипидемияСемейная гиперхолестеринемия относится к группе метаболических нарушений, получивших название гиперлипопротеинемий, характеризующихся высокими уровнями липидов (холестерина, триглицеридов, или и того и другого сразу) и специфических липопротеинов плазмы. Описаны также и другие моногенные гиперлипопротеинемий, каждая с четким биохимическим и клиническим фенотипом. Оказалось, что, кроме мутаций в гене рецептора ЛПНП, к семейной гиперхолестеринемии также ведут аномалии в трех других генах. Примечательно, что все четыре гена, ответственные за семейную гиперхолестеринемию, нарушают либо функцию или количество рецептора ЛПНП в правильном положении на поверхности клетки, либо апопротеин В-100, белковый компонент лиганда ЛПНП-рецептора. Неудивительно, что клинические фенотипы индивидуумов, несущих мутации в этих четырех генах, трудноразличимы. Вследствие ее значимости рассмотрим семейную гиперхолестеринемию, вызванную мутациями в рецепторе ЛПНП. Мы обсудим также мутации в гене протеазы PCSK9; хотя некоторые мутации в этом гене вызывают гиперхолестеринемию, большее значение PCSK9 в том, что ее некоторые частые варианты понижают уровень ЛПНП-холестерина плазмы в общей популяции, защищая от заболевания коронарных сосудов сердца. Семейная гиперхолестеринемия вследствие мутаций в рецепторе ЛПНПМутации в гене, кодирующем рецептор ЛПНП, — самая частая причина семейной гиперхолестеринемии. Рецептор представляет собой поверхностный клеточный белок, ответственный за связывание ЛПНП и доставку его внутрь клетки. Как у гетерозигот, так и у гомозигот развивается ранняя патология сердца в результате появления атером (отложения производных холестерина и ЛПНП в венечных артериях), ксантом (холестерин депонируется в коже и сухожилиях) и старческой дуги (отложение холестерина вокруг периферии роговицы). Так тщательно пока изучены немногие болезни; полностью подтверждена вся последовательность патологических событий от мутантного локуса до его эффекта на уровне человека и популяции.
Генетика семейной гиперхолестеринемии вследствие мутаций в рецепторе ЛПНП. Семейная гиперхолестеринемия, вызванная мутациями в рецепторе ЛПНП, наследуется как аутосомный полудоминирующий признак. Известны как гомозиготные, так и гетерозиготные фенотипы, очевиден эффект дозы генов; болезнь обнаруживается раньше и проявляется сильнее у гомозигот, чем у гетерозигот, отражая большее снижение числа рецепторов ЛПНП и большее повышение ЛПНП в плазме. Гомозиготы могут уже в детстве иметь клинически значимое заболевание коронарных сосудов сердца, немногие доживают до 40 лет. Гетерозиготная форма болезни с популяционной частотой около 1 на 500 — одно из наиболее частых моногенных заболеваний человека. Гетерозиготы имеют высокие уровни холестерина плазмы, почти в 2 раза выше, чем в контрольной группе. По причине наследственной природы семейной гиперхолестеринемии важно подтвердить или отвергнуть этот диагноз, встречающийся приблизительно у 5% выживших после инфаркта миокарда гетерозигот по дефекту рецептора ЛПНП. В то же время семейную гиперхолестеринемию имеет только около 1 из 20 в общей популяции лиц с повышенным холестерином и гиперлипопротеинемией, аналогичной имеющейся при гетерозиготной недостаточности рецептора ЛПНП, чаще такие индивидуумы имеют гиперхолестеринемию многофакторного происхождения. Захват холестерина рецептором ЛПНП. Нормальные клетки получают холестерин, или синтезируя его, или захватывая из плазмы внешний холестерин, связанный с ЛПНП. Захват осуществляет рецептор ЛПНП, распознающий апопротеин В-100, белковый остаток ЛПНП. Рецепторы ЛПНП на поверхности клетки локализуются в углублениях (закрытых ямках), создаваемых белком клатрином. Связанный рецептором ЛПНП переносится в клетку за счет эндоцитоза ямок, которые в конце концов попадают в лизосомы, где ЛПНП гидролизуются, освобождая холестерин. Увеличение свободного внутриклеточного холестерина уменьшает внутренний синтез холестерина, подавляя ограничивающий фермент пути синтеза (3-гидрокси-З-метилглутарил-коА-редуктазу). Холестерин, ненужный для клеточного метаболизма или синтеза мембран, может реэтерифицироваться и накапливаться в виде эфиров холестерина. Этот процесс стимулируется активацией ацетилкоэнзим-А-холестерол-ацилтрансферазы. Увеличение внутриклеточного холестерина также уменьшает синтез рецептора. Классы мутаций в рецепторе ЛПНП. В гене рецептора ЛПНП обнаружено более 700 различных мутаций, распределенных по всей его длине. (Часто остается неясным, все ли эти варианты нуклеотидной последовательности патогенны или некоторые из них — варианты нормы без фенотипического проявления.) Подавляющее большинство аллелей — однонуклеотидные замены, небольшие инсерции или делеции; структурные перегруппировки занимают в большинстве популяций всего от 2-10% аллелей рецептора ЛПНП. Зрелый рецептор ЛПНП имеет пять четких структурных областей, в основном имеющих различающиеся функции. В определении функций этих областей заметную роль сыграл анализ эффекта мутаций в каждой области рецептора. Эти исследования демонстрируют тот вклад, который может внести генетический анализ в определение связей между структурой и функциями белков. Для исследования мутантных рецепторов и получающихся сбоев в клеточном метаболизме холестерина проведены исследования в культурах фибробластов больных. Мутации в гене рецептора ЛПНП можно сгруппировать в шесть классов, в зависимости от нарушаемого мутацией этапа нормального клеточного цикла рецептора. Мутации 1-го класса — аллели, полностью нарушающие синтез рецептора, наиболее частый тип патогенных мутаций в этом локусе. В остальных пяти классах рецептор синтезируется нормально, но нарушены его функции. Мутации в классах 2, 4 и 6 определяют характеристики полипептида, критичные для его субклеточной локализации. Сравнительно частые мутации 2-го класса нарушают перенос рецепторов ЛПНП, и они накапливаются в месте их синтеза — эндоплазматическом ретикулуме вместо перемещения в комплекс Гольджи. Эти аллели нарушают соответствующую третичную структуру белка, необходимую для его выхода из эндоплазматического ретикулума. Мутантные рецепторы 3-го класса достигают поверхности клетки, но не способны связывать ЛПНП. Таким образом, эти аллели позволили исследователям идентифицировать область связывания ЛПНП. Мутации 4-го класса нарушают прикрепление рецептора в клатриновой ямке и связанный ЛПНП не переносится в клетку. Данные мутации изменяют или удаляют цитоплазматическую область в карбоксильном конце рецептора, доказывая, что этот участок в норме направляет рецептор в ямку. Мутации 5-го класса — аллели, нарушающие восстановление рецептора. Для восстановления рецептора необходима его диссоциация со связанным ЛПНП в эндосоме. Мутации в гомологичном участке предшественника фактора роста эпидермиса нарушают освобождение лиганда ЛПНП. Это приводит к распаду рецептора, возможно, вследствие того, что занятый рецептор не может вернуться на поверхность клетки. Протеазы PCSK9 и их отношения с ЛПНП и холестерином при гиперхолестеринемииМиссенс-мутации с увеличением функции в гене, кодирующем протеазу PCSK9, оказались причиной редкой аутосомно-доминантной семейной гиперхолестеринемии. Эксперименты показывают, что повышенная активность протеазы PCSK9 ведет к распаду рецептора ЛПНП (хотя неясно, является ли рецептор ее прямой мишенью), регулируя тем самым уровень рецептора в гепатоцитах. Следовательно, протеаза действует как механизм, уменьшающий количество молекул рецептора и предохраняющий от чрезмерного захвата холестерина, что необходимо, например, в случае низкохолестериновой диеты. Миссенс-мутации в протеазах PCSK9, связанных семейной гиперхолестеринемией, оказывается, вызывают болезнь, увеличивая активность протеаз, тем самым снижая содержание рецептора ЛПНП до аномально низкого уровня. Мутации в гене PCSK9 с увеличением функции, вызывающие семейную гиперхолестеринемию, показали, что протеазы PCSK9 — основной регулятор метаболизма холестерина ЛПНП. Защитное действие некоторых вариантов нуклеотидной последовательности гена PCSK9 от ишемической болезни сердца Связь между семейной гиперхолестеринемией и геном PCSK9 позволяет предполагать, что частые варианты нуклеотидной последовательности в гене PCSK9 могут быть связаны как с очень высоким, так и очень низким уровнем холестерина ЛПНП в общей популяции (несмотря на то что, к сожалению, для частых вариантов в других генах, включая три других, связанных с семейной гиперхолестеринемией, связь с изменениями уровня холестерина плазмы в общей популяции убедительно не доказана). Обнаружено несколько вариантов последовательности гена PCSK9, тесно связанных с низким уровнем холестерина ЛПНП в плазме. Например, в афроамериканской популяции с очень низким уровнем холестерина ЛПНП один из двух нонсенс-вариантов PCSK9 обнаруживается в 2,6% всех случаев; присутствие любого варианта приводит к уменьшению уровня холестерина ЛПНП на 40%. Это снижение холестерина ЛПНП оказывает мощный защитный эффект против ИБС, уменьшающий ее риск почти на 90%; только около 1% чернокожих носителей любого из двух вариантов нонсенс-мутаций за 15-летний период наблюдения имели ИБС, по сравнению с почти 10% без любой из этих двух мутаций. Другой аллель (Arg46Leu) — более частый (3,2%) у европеоидов и вызывает только 15% уменьшение уровня холестерина ЛПНП, но, что удивительно, на 50% уменьшает заболеваемость ИБС. Эти сведения имеют большое значение для здравоохранения, поскольку они позволяют предполагать, что незначительное, но продолжающееся всю жизнь уменьшение холестерина ЛПНП плазмы на 20-40 мг% должно значительно уменьшить встречаемость ИБС в популяции. Наконец, эти открытия показывают, как исследование редкого генетического нарушения может привести к важному новому знанию о генетическом вкладе в частые генетически комплексные болезни. Патогенез атеросклеротических бляшек при семейной гиперхолестеринемииНесмотря на более чем 30-летний период исследований, приведший к впечатляющему расширению знаний биологии рецептора ЛПНП и семейства молекулярных дефектов, влияющих на развитие семейной гиперхолестеринемии, механизмы, которыми повышение уровня ЛПНП приводит к образованию атеросклеротических бляшек в артериях, все еще не ясны. У гомозигот повышенный уровень ЛПНП снижается во внеклеточной жидкости альтернативными рецепторами — «падалыциками» некоторых клеток, например макрофагов. Исследования макрофагов in vitro показывают, что избыточный холестерин хранится в виде микрокапель эфиров холестерина, вызывая появление пенистых клеток, обычно обнаруживаемых в ксантомах и атеросклеротических бляшках, но значимость этого процесса in vivo в настоящее время не определена. Наконец, выяснение биохимической основы семейной гиперхолестеринемии оказало глубокое влияние на лечение значительно более частых форм спорадической гиперхолестеринемии, приведя к созданию статинового класса лекарств, тормозящих биосинтез холестерина. Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021 — Также рекомендуем «Генетика муковисцидоза. Наследование» Оглавление темы «Генетика заболеваний»:
|
Источник