Замена нуклеотида в гене гемоглобина
Содержание статьи
уклеотидные замены: миссенс-мутации
Нуклеотидные замены: миссенс-мутации
Замена единичного нуклеотида (или точковая мутация) в последовательности ДНК может изменять код в триплете и вызывать замену одной аминокислоты другой в продукте гена. Такие мутации называются миссенс-мутациями, поскольку они изменяют значение кодирующей последовательности гена, определяя другую аминокислоту.
При многих заболеваниях, например гемоглобинопатиях, наиболее часто обнаруживаемые мутации — миссенс-мутации.
Другие замены нуклеотидов, происходящие как в пределах, так и за пределами кодирующей последовательности гена, также могут влиять на продукт гена или создавать помехи непосредственно самому процессу транскрипции.
Множество мутаций в 5′-области промотора или 3′-нетранслируемой области гена b-глобина ведет к выраженному уменьшению количества готовой, зрелой мРНК b-глобина. На самом деле такие мутации позволили объяснить значение для экспрессии генов конкретных нуклеотидов в этих областях.
Мутации прекращающие синтез цепи
Точковые мутации последовательности ДНК, вызывающие замены нормального кодона аминокислоты на один из трех стоп-кодонов, называются нонсенс-мутациями. Поскольку при достижении стоп-кодона трансляция мРНК прекращается, мутации, преобразующие один из кодирующих кодонов экзона в стоп-кодон, прекращают на полпути трансляцию кодирующей последовательности мРНК.
Последствия мутаций преждевременного завершения трансляции двойственны. Во-первых, мРНК, несущая такую мутацию, часто неустойчива (нонсенс-распад мРНК), и трансляция оказывается невозможной. Даже если полученная мРНК достаточно стабильна для трансляции, усеченный белок обычно также неустойчив и быстро деградирует в пределах клетки.
Точковая мутация может не только создать кодон преждевременного завершения трансляции, но и уничтожить стоп-кодон, позволив продолжение трансляции до следующего стоп-кодона. Такая мутация создает белок с дополнительными аминокислотами на карбоксильном конце и может нарушать любые управляющие функции, предусматриваемые 3′-нетранслируемым участком, расположенным ниже нормального стоп-кодона.
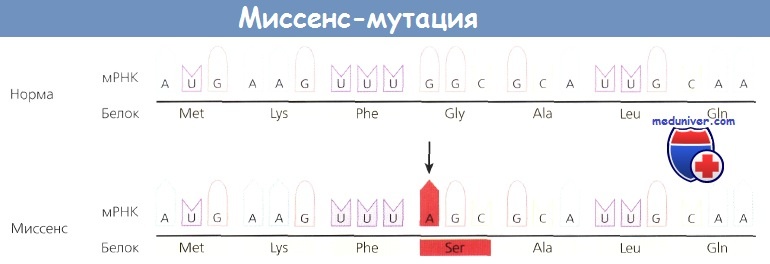
Миссенс-мутация — замена на кодон, кодирующий другую аминокислоту (может быть опасной мутацией или нейтральным полиморфизмом)
Мутации процессинга РНК
Нормальный механизм преобразования первичной РНК в зрелую мРНК требует серии модификаций, включая полиаде-нилирование, закрытие 5′-конца и сплайсинг. Созревание РНК зависит от специфических последовательностей в пределах мРНК. При сплайсинге описаны два основных класса мутаций. Чтобы получить зрелую мРНК удалением интронов и сращиванием экзонов, необходимы конкретные нуклеотидные последовательности, располагающиеся непосредственно в или около соединений экзона и интрона (5′-донорский сайт) или интрона и экзона (3′-акцепторный сайт).
Мутации, повреждающие необходимые нуклеотиды в донорском и акцепторном сайте, нарушают (и в некоторых случаях прекращают) нормальный сплайсинг РНК в этом месте.
Второй класс мутаций сплайсинга включает замены оснований в интроне, не влияющие на донорский или акцепторный сайты. Данный класс мутаций создает альтернативный донорский или акцепторный сайт, конкурирующий с нормальными сайтами в ходе сплайсинга РНК. Таким образом, в этих случаях по крайней мере часть зрелой мРНК может иметь неправильно сращенные последовательности интрона.
Горячие точки мутаций
Нуклеотидные замены, включающие замену одного пуринового основания другим (А на G или наоборот) или одного пиримидинового на другое пиримидиновое (С на Т или Т на С), называются транзициями. Замена же пуринового основания на пиримидиновое (или наоборот) называется трансверсией. Если бы нуклеотидные замены были случайными, должно было быть в два раза больше трансверсий, чем транзиций, поскольку каждое основание может подвергаться двум трансверсиям и только одной транзиций. Разные мутагенные процессы преимущественно вызывают тот или другой тип замены.
Например, среди однонуклеотидных замен, вызывающих генетические болезни, преимущественно бывают транзиций. Это наблюдение, вероятно, может объясняться тем, что основная форма модификации ДНК в геноме человека представлена метилированием остатка цитозина (с формированием 5-метилцитозина), особенно когда он располагается рядом с гуанином (т.е. как динуклеотид 5′-CG-3′).
Спонтанное деаминирование 5-метилцитозина в тимидин в CG-динуклеотиде вызывает транзицию С > Т или G > А (в зависимости от нити ДНК, в которой видоизменяется 5-метилцитозин). Более 30% всех однонуклеотидных замен относятся к этому типу, они происходят в 25 раз чаще любой другой однонуклеотидной мутации. Таким образом, CG-динуклеотид представляет настоящую «горячую точку» для мутаций в геноме человека.
— Также рекомендуем «Делеции и инсерции: виды, последствия»
Оглавление темы «Мутации»:
- Виды мутаций у человека. Варианты
- Происхождение мутаций: геномные, хромосомные, генные мутации
- Нуклеотидные замены: миссенс-мутации
- Делеции и инсерции: виды, последствия
- Динамические мутации. Оценка частоты мутаций
- Номенклатура мутаций. Принятые обозначения
- Половые различия в частоте мутаций
- Генетический полиморфизм. Геномика персонализированной медицины
- Полиморфизм ДНК: варианты
- Генетика группы крови и их полиморфизмы
Источник
енетика серповидноклеточной анемии. Наследование
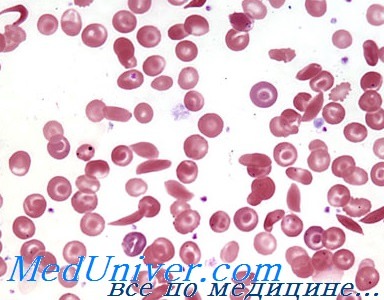
Генетика серповидноклеточной анемии. НаследованиеHbS был первым обнаруженным аномальным гемоглобином с высоким клиническим значением. Он возникает вследствие замены единственного нуклеотида, которая изменяет кодон шестой аминокислоты В-глобина глутаминовой кислоты на валин (GAG -> GTG: Glu6Val). Гомозиготность по данной мутации — причина серповидноклеточной анемии, серьезного заболевания, часто встречающегося в некоторых частях света. Болезнь имеет характерное географическое распределение, чаще всего встречается в Экваториальной Африке и реже всего в Средиземноморье, Индии и странах, в которые мигрировали люди из этих регионов. С этой, обычно фатальной в раннем детстве болезнью рождаются около 1 из 600 афроамериканцев, хотя все более частым становится более долгое выживание. Серповидноклеточная анемия — тяжелое аутосомно-рецессивное гемолитическое заболевание, характеризующееся тенденцией эритроцитов принимать выраженно аномальную форму (серпа) в условиях низкого насыщения кислородом. Гетерозиготы, про которых говорят, что они имеют «признак» серповидноклеточно-сти, обычно клинически здоровы, но их эритроциты в условиях очень низкого давления кислорода in vitro принимают форму серпа. Случаи, когда бы это могло происходить in vivo, редки, хотя гетерозиготы имеют риск инфаркта селезенки, особенно при полетах на большой высоте в самолетах с низким давлением в кабине. Гетерозиготное состояние наблюдают приблизительно у 8% афроамериканцев, но в областях, где частота гена высокая (например, в Западной Центральной Африке), вплоть до 25% новорожденных — гетерозиготы. Молекулярная патология HbS — серповидноклеточной анемииОколо 50 лет тому назад Ингрэм обнаружил, что аномалия HbS связана с заменой одной из 146 аминокислот в В-цепи молекулы гемоглобина. Все клинические проявления наличия HbS — последствия этого единственного изменения в гене В-глобина. Это было первой демонстрацией того, что мутация в структурном гене может вызывать замену аминокислоты в соответствующем белке. Поскольку аномалия HbS локализуется в В-цепи, формула HbS может быть записана как а2b2s или, более точно, а2Ab2s.
Гетерозиготы имеют смесь двух типов гемоглобинов (НbА и HbS), обозначаемых а2Аb2А, а2Аbs, а также гибридный тетрамер гемоглобина, обозначаемый как a2AbA,bs. Серповидноклеточность и ее последствияМолекулы гемоглобина, содержащие мутантные субъединицы b-глобина, нормальны по их способности выполнять их главную функцию связывания кислорода (если они не полимеризованы, как указано далее), но в ненасыщенной кислородом крови они растворимы в пять раз меньше по сравнению с нормальным гемоглобином. Относительная нерастворимость дезоксигемоглобина S является физической основой феномена серповидноклеточности. В условиях низкой кислородной напряженности молекулы HbS собираются в форме полимеров, формирующих стержни или волокна, искажающие форму эритроцитов. Эти уродливые эритроциты деформируются хуже, чем в норме, и, в отличие от нормальных красных кровяных клеток, не могут сжиматься, проходя через капилляры, тем самым блокируя ток крови и вызывая локальную ишемию. Происхождение мутаций гемоглобина SУ большинства лиц африканского происхождения нормальный ген b-глобина содержится в пределах фрагмента рестрикции размером в 7,6 килобазы ДНК. В то же время в определенных частях Африки, например в Гане и почти у 70% афроамериканцев, аллель серповидноклеточного глобина часто обнаруживают во фрагменте размером в 13 килобаз. Частая ассоциация серповидноклеточного глобина с 13-килобазовым фрагментом — поразительный пример неравновесного сцепления. В других частях Африки (например, в Кении) мутация серповидноклеточности обычно связана с фрагментом размером в 7,6 килобазы. Эти находки позволяют утверждать, что мутация серповидноклеточности возникла в Западной Африке в хромосоме, которая содержала ген р-глобина во фрагменте длиной 13 килобаз, и что подобная мутация, по крайней мере, один раз, независимо произошла где-то еще. Защита от малярии, обеспечиваемая данной мутацией у гетерозигот, обеспечила ее высокую частоту в областях, пораженных малярией. — Также рекомендуем «Генетика гемоглобинов HbС, Hammersmith. Наследование» Оглавление темы «Генетика гемоглобинопатий»:
|
Источник
арианты патологического гемоглобина. Гемоглобинопатии
Варианты патологического гемоглобина. Гемоглобинопатии
Наследственные нарушения гемоглобина можно подразделить на три большие группы, в зависимости от того, изменяет мутация белок глобина, его синтез или переключение глобинов:
• структурные варианты, изменяющие полипептид глобина, не влияя на показатели синтеза;
• талассемии, когда снижается синтез (или, редко, имеется крайняя нестабильность) одной или более цепей глобина, что приводит к дисбалансу относительных количеств а- и b-цепей;
• наследственное персистирование фетального гемоглобина, группа клинически благоприятных состоянии, представляющих интерес, поскольку они нарушают внутриутробное переключение синтеза от у-глобина к b-глобину.
Различные гемоглобины произошли вследствие точковых мутаций в одном из структурных генов глобина, но некоторые сформировались благодаря другим, более сложным молекулярным механизмам. Описано более 400 аномальных гемоглобинов, приблизительно половина их клинически значима.
Структурные варианты гемоглобина можно разделить на три класса, в зависимости от клинического фенотипа.
• Варианты, вызывающие гемолитическую анемию. Подавляющее большинство мутантных гемоглобинов, вызывающих гемолитическую анемию, приводят к неустойчивости тетрамера гемоглобина. Тем не менее два самых известных варианта, связанных с гемолизом, серповидно-клеточный гемоглобин (HbS) и НbС, не связаны с неустойчивостью, а заставляют мутантные белки глобина принимать необычную ригидную форму.
• Варианты с измененным транспортом кислорода вследствие повышенного или сниженного сродства к кислороду или образования метгемоглобина, формы глобина, неспособного к обратимому обогащению кислородом.
• Варианты, вызванные мутациями в кодирующем регионе, приводящими к талассемиям, за счет уменьшения количества полипептида глобина. Большинство этих мутаций нарушает скорость синтеза мРНК или белка. Некоторые редкие варианты вызывают общую нестабильность мономера гемоглобина, большую, чем при вариантах, ведущих к гемолитической анемии.
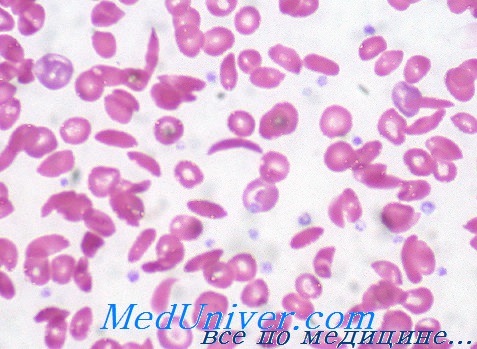
Структурные изменения выбраны для описания в данной статье, либо потому что они часто встречаются и типичны для одной из трех групп, либо иллюстрируют драматические биохимические и клинические последствия мутаций.
Варианты патологического гемоглобина:
1. HbS. Замена единственного нуклеотида. b-цепь: Glu6Val. Полимеризация ненасыщенного кислородом HbS -> серповидноклеточность —> закупорка сосудов и гемолиз. Аутосомно-рецессивное наследование.
2. HbC. Замена единственного нуклеотида. b-цепь: Glu6Lys. Насыщенный кислородом НbС стремится кристаллизоваться -> меньшая деформация клеток —> легкий гемолиз. Аутосомно-рецессивное наследование. Болезнь у компаундных гетерозигот HbS/HbC напоминает легкую серповидноклеточную анемию
3. Гемоглобин Hammersmith. Замена единственного нуклеотида. b-цепь: Phe42Ser. Неустойчивый гемоглобин -> преципитация гемоглобина —> гемолиз; также снижение сродства к кислороду. Аутосомно-доминантное наследование.
4. Hyde Park HbM. Замена единственного нуклеотида. b-цепь: His92Tyr. Замена делает окисленное железо гема устойчивым к редуктазе метгемоглобина —>HbM, не способный переносить кислород ->цианоз (бессимптомный). Аутосомно-доминантное наследование.
5. Гемоглобин Kempsey. Замена единственного нуклеотида. b-цепь: Asp99Asn. Замена удерживает гемоглобин в форме высокого сродства к кислороду —> снижение кислорода в тканях —> полицитемия. Аутосомно-доминантное наследование.
6. HbЕ. Замена единственного нуклеотида. b-цепь: Glu26Lys. Мутация —> аномальная структура гемоглобина и уменьшение его синтеза (аномальный сплайсинг РНК) —> мягкая талассемия. Аутосомно-рецессивное наследование.
— Также рекомендуем «Генетика серповидноклеточной анемии. Наследование»
Оглавление темы «Генетика гемоглобинопатий»:
- Варианты патологического гемоглобина. Гемоглобинопатии
- Генетика серповидноклеточной анемии. Наследование
- Генетика гемоглобинов HbС, Hammersmith. Наследование
- Генетика гемоглобинов Hyde Park, Kempsey. Наследование
- Генетика альфа-талассемии. Наследование
- Генетика бета-талассемии. Наследование
- Генетика сложных талассемий. Наследование
- Профилактика талассемии. Скрининг
- Болезни из-за мутации белков. Примеры
- Генетическая гетерогенность: аллельная, локусная, гены-модификаторы
Источник
енетика бета-талассемии. Наследование
Генетика бета-талассемии. Наследованиеb-Талассемии имеют много общих характеристик с а-талассемией. Уменьшение синтеза b-глобина вызывает гипохромную микроцитарную анемию, а дисбаланс в синтезе глобинов ведет к осаждению избыточных а-цепей, вызывая патологию мембраны эритроцитов. В отличие от а-глобина, b-цепь важна только в послеродовом периоде. Следовательно, В-талассемии не проявляются несколько месяцев после рождения, когда b-глобин в норме заменяет у-глобин как основную не-a-цепь, и начинается синтез только основного гемоглобина взрослых, НbА. Избыточные а-цепи накапливаются в эритроцитах и их предшественниках, приведя к их гибели и нарушению эритропоэза. Поскольку 6-ген остается неповрежденным, производство НbА2 продолжается, и, фактически, повышение уровня НbА2 уникально для гетерозиготных носителей b-талассемии. Уровень HbF также повышен, но не потому, что вновь активизировалась экспрессия гена у-глобина, отключенная при рождении, а из-за выборочного выживания или возможного повышения производства незначительной популяции эритроцитов взрослых, содержащих HbF. В отличие от a-талассемий, b-талассемии обычно вызываются однонуклеотидными заменами, а не делециями. Во многих регионах мира, где часто встречается b-талассемия, имеется много разных мутаций гена b-талассемии, и наиболее вероятно, что лица, несущие два аллеля b-талассемии, окажутся генетическими компаундами, а не истинными гомозиготами по одному аллелю. Большинство больных с двумя аллелями b-талассемии имеют «большую» талассемию, заболевание, проявляющееся выраженной анемией и пожизненной потребностью в медицинском наблюдении. Когда аллели b-талассемии допускают настолько малый синтез b-глобина, что гемоглобин полностью отсутствует, заболевание определяют как b0-талассемию. Если какое-то количество гемоглобина обнаруживается, говорят, что у пациента имеется b+-талассемия. Хотя тяжесть клинических проявлений болезни зависит от комбинированного эффекта двух имеющихся аллелей, до недавних пор доживание до взрослого возраста было редкостью. У детей, гомозиготных по b-талассемии, анемия развивается сразу же, как только уменьшается синтез HbF, обычно до 2 лет жизни. Эритроциты периферической крови заметно гипохромны и имеют различную величину и форму. В настоящее время лечение талассемий основано на коррекции анемии и стимуляции костного мозга переливаниями крови и стимуляции накопления железа назначением хе-латов. Пересадка костного мозга эффективна, но ее применяют только при наличии члена семьи с соответствующим HLA-генотипом. Носители только одного аллеля b-талассемии клинически здоровы, такую форму заболевания называют «малой». Характерны гипохромные микроцитарные эритроциты, возможна легкая анемия, которая может быть диагностирована как железодефицитная. Диагноз малой талассемий подтверждается электрофорезом гемоглобинов, обычно показывающим увеличение уровня HbA2 (a2q2).
b-Талассемия, сложные талассемий и наследственное персистирование фетального гемоглобинаПочти все известные типы мутаций, уменьшающих синтез мРНК или белка, описаны как причина b-талассемии. Таким образом, приведенный ниже обзор генетических дефектов, описывающих конкретные молекулярные основы одной из наиболее частых и тяжелых генетических болезней в мире, полезен для общего понимания мутационных механизмов. Мутации комплекса b-глобина подразделяют на две обширные группы с разными клиническими фенотипами. Одна группа дефектов, характерная для подавляющего большинства пациентов, нарушает синтез только В-глобина, вызывая простую b-талассемию. Во второй группе мутаций большие делеции вызывают сложные талассемий, в которых утрачивается ген b-глобина, а также один или более других генов (или LCR) в группе b-глобина. Некоторые делеции в пределах группы b-глобина вызывают не талассемию, а довольно интересный фенотип, характеризующийся НПФГ (т.е. продолжение экспрессии гена у-глобина во взрослой жизни). Молекулярная основа простой b-талассемииПростая b-талассемия возникает вследствие множества различных типов молекулярных аномалий, преимущественно точковых мутаций, в гене b-глобина. Единственная частая делеция b-глобина среди всех расовых групп — 619-bр, частичная делеция 3′-конца гена у пациентов азиатского (индийского) происхождения. Большинство мутаций, вызывающих простую b-талассемию, приводят к уменьшению количества синтезируемой мРНК b-глобина. Они включают мутации промотора, мутации сплайсинга РНК (наиболее частые), мутации, нарушающие фланкирование мРНК, и мутации сдвига рамки или нонсенс-мутации, вводящие кодон преждевременного завершения в пределах кодирующей области гена. Несколько структурных вариантов гемоглобина также нарушают процессинг мРНК b-глобина, например HbЕ, описанный ниже.
Мутации сплайсинга РНК при бета-талассемииБольшинство пациентов с b-талассемиями, вызванными уменьшением синтеза мРНК b-глобина, имеют аномалии сплайсинга РНК. Описано более двух десятков дефектов этого типа, и, вместе взятые, они имеют большое клиническое значение. Эти мутации стали широко известны, поскольку их влияние на сплайсинг часто оказывалось неожиданно сложным, и анализ мутантных мРНК содействовал познанию критической последовательности нормального процессинга РНК. Дефекты сплайсинга разделяют на три группы, в зависимости от региона непроцессируемой РНК, в котором располагается мутация. Группа 1, мутации сайта сплайсинга включает мутации в 5′-донорском или 3′-акцепторном участках интронов или в окружающих управляющих последовательностях. Критическая роль консервативных динуклеотидов GT в 5′-донорском участке и AG в З’-акцепторном участках интрона следует из полной утраты нормального сплайсинга, вызванной мутациями этих динуклеотидов. Инактивация нормального сайта акцептора приводит к использованию других последовательностей в исходной РНК, сходных с акцептором. Такие альтернативные сайты называют криптическими, поскольку в норме, при наличии правильного сайта, они не используются при сплайсинге. Криптические донорские или акцепторные сайты сплайсинга могут быть как в интронах, так и в экзонах, и могут использоваться в конкуренции с другими криптическими и нормальными сайтами сплайсинга. Значение согласованных последовательностей, смежных с донорским или акцепторным динуклеотидами, также проявляется по эффектам мутаций. Например, замена пятого или шестого нуклеотида донорской последовательности интрона 1 уменьшает эффективность нормального сплайсинга, но, поскольку некоторый объем нормального сплайсинга все же происходит, формируется фенотип b+-талассемии. Группа 2, мутации интрона — результат мутаций в пределах криптического сайта сплайсинга в интроне, повышающих использование этого сайта, что делает его более сходным или даже идентичным нормальному сайту сплайсинга. Затем пусковой криптический сайт с переменной эффективностью конкурирует с нормальным сайтом, уменьшая количество нормальной мРНК за счет нарушения сплайсинга в «правильном» сайте, остающемся вполне сохранным. Мутации криптических сайтов сплайсинга часто «неполные». Это означает, что происходит определенный объем сплайсинга с использованием нормального сайта, приводя к фенотипу b+-талассемии. Группа 3, изменения кодирущей последовательности, влияющие на сплайсинг, вызываются мутациями сдвига рамки, которые активируют криптический сайт сплайсинга в экзоне. Например, одна из легких форм b+-талассемии вызвана мутацией в кодоне 24, активизирующей криптический сайт сплайсинга, но не изменяющей закодированную аминокислоту [как GGT, так и GGA кодируют глицин]; это пример синонимичной мутации, оказавшейся не безразличной по своему эффекту. Структурный вариант НbЕ демонстрирует, как нарушение сплайсинга и изменение в кодирующей последовательности могут вызываться единственной мутацией. Нефункциональная мРНК при бета-талассемииНекоторые синтезированные молекулы мРНК нефункциональны и не могут управлять синтезом полного полипептида, поскольку мутация генерирует преждевременные стоп-кодоны, раньше времени завершающие трансляцию белка. Две мутации около аминового конца поясняют этот эффект при b-талассемии. При одной (Gln39Stop) нарушение трансляции — следствие замены единственного нуклеотида, создающей нонсенс-мутацию. Другая мутация типа сдвига рамки происходит вследствие делеции одной пары оснований в начале открытой рамки считывания, с потерей первого нуклеотида 16 кодона, в норме кодирующего глицин; при произошедшем сдвиге рамки почти сразу образуется стоп-кодон, прерывающий дальнейшее считывание. Поскольку синтез b-глобина отсутствует, оба типа нефункциональных мутаций мРНК вызывают b°-талассемию. В отличие от этого, мутации сдвига рамки, расположенные около карбоксильного конца белка, позволяют большой части мРНК транслироваться нормально или вызывают удлинение цепей глобина, чаще приводя к различным вариантам гемоглобина, а не к b°-талассемии. Кроме снижения синтеза полипептида b-глобина, нонсенс-кодоны, включая два вышеописанных, часто приводят к уменьшению количества мутантной мРНК, которая на самом деле может не обнаруживаться. Механизмы, лежащие в основе этого феномена, названного нонсенс-опосредованным разрушением мРНК, не полностью понятны, но эффект ограничен нонсенс-кодонами, расположенными далее 50 пар оснований в 5′-конце последнего соединения между экзонами. Дефекты фланкирующих последовательностей мРНК бета-глобина при бета-талассемииДве мутации при b+-талассемии указывают на критическую роль посттранскрипционных модификаций любой мРНК, кэпирования РНК, ограничивающее 5′-конец, и полиаденилирования 3-конца мРНК. Обнаружен один пациент с заменой аденина на цитозин в первом нуклеотиде мРНК (это место — сайт кэпирования — в 90% эукариотической мРНК — пурин). Мутация приводит к нарушению формирования кэпа, вызывая распад РНК. Полиаденилирование мРНК происходит после ферментативного расщепления, а сигнал точки расщепления, AAUAAA, находится около 3′-конца в большинстве эукариотической мРНК. Пациент с мутацией, изменяющей сигнальную последовательность на ААСААА, синтезировал только незначительное количество мРНК b-глобина, полиаденилированной в нормальном положении. — Также рекомендуем «Генетика сложных талассемий. Наследование» Оглавление темы «Генетика гемоглобинопатий»:
|


Источник